THE SILENT TRANSITIONS IN CANCER RESEARCH
Changes in scientific paradigms tend to be incubated silently in the shadow of conventional ideas and practice. This is what is happening in cancer research.
For decades, cancer research has aimed to find a cure based on complete elimination of the malignant cell population, while damaging normal cells as little as possible. This is more feasible with surgery, radiotherapy and chemotherapy in early stages of the disease; and therefore screening programs with appropriate diagnostic techniques are expected to reach upstream to detect cancers as early as possible. Most guidelines for national cancer programs—in addition to recommending primary prevention—follow this paradigm.[1,2] The reasoning is that effective screening should increase the proportion of disease detected at curable stages and therefore decrease mortality rates.
Such expectations seemed to be confirmed by a modest but consistent decrease in age-adjusted cancer mortality rates in the USA and Europe over in the last two decades.[3,4]
Additionally, rapidly improving techniques for DNA sequencing allowed identification of mutations in cancer cells, providing potential targets for specific drugs, some of which induce impressive responses in patients carrying the mutated gene.[5]
At the same time, new evidence and concepts started to appear, pointing in another direction.
- Increasing prevalence The number of people living with cancer is increasing and will continue to increase, as shown by crude (not age-adjusted) rates. This is mainly due to population aging and increasing survival rates.[6]
- The notion of chronicity Improvements in treatment of advanced cancer imply more years with better quality of life, the disease becoming controllable, if not curable.[7,8] This in turn contributes to increasing prevalence.
- Limitations of population screening programs While cervical cancer screening has had a major impact on mortality, the results of mammography screening for breast cancer and colonoscopy screening for colorectal cancer, while statistically significant, have not had a substantial impact on mortality. The results of tomography screening for lung cancer are still being debated, and population screening with PSA for prostatic cancer is currently not recommended.[9,10]
- The short span of cancer remission with targeted therapies Cancer cell mutation rates and the redundancy of molecular loops controlling cell proliferation entail rapid appearance of resistance, thus preventing short-term remission from translating into improved survival.[11]
- The re-emergence of immunotherapy, this time with drugs targeting the immune system, not the tumor itself.
This new evidence and conceptual transitions point to a future in which the medical challenge will be chronic treatment options for an increasing population of patients with tumors that are not curable, but controllable. What do we have in our arsenal to address this challenge?
THE ACCUMULATED KNOWLEDGE OF FUNDAMENTAL TUMOR IMMUNOLOGY IN THE 21st CENTURY
Immunotherapy will have a major role in the new scenario. Intense exploration of the notion of mobilizing the immune system to control cancer cell proliferation started 50 years ago, but failed to fulfill its promise in the 20th century. We entered the 21st century with interferon producing minor effects in a few neoplasms (hairy cell leukemia being the one tumor to react most favorably), with only one monoclonal antibody registered for cancer (rituximab for CD20 lymphoma) and no therapeutic vaccine in clinical practice.
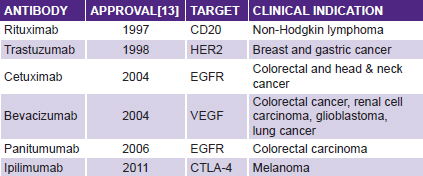
But there are reasons for fresh optimism in cancer immunotherapy: first, monoclonal antibodies are one of the greatest successes in cancer therapy over the last decade. There are now a dozen monoclonal antibodies showing antitumor effects in clinical settings.[12] The most commonly used are shown in the table below, and another 200 are estimated in different phases of development.[13]
The first antibodies showing antitumor activity were directed at targets on cancer cells. A major step forward occurred in 2010, with the report of antitumor activity in advanced melanoma by ipilimumab. This antibody is not aimed at cancer cells, but targets the immune system itself, blocking CTLA-4, a molecule that mediates down-regulation of the immune response.[14] This finding made cancer immunotherapy the medical breakthrough of 2013.[15]
In Cuba, the humanized monoclonal antibody nimotuzumab, which targets the epidermal growth factor receptor (EGFR) in cancer cells, is registered for head and neck, brain, and esophageal tumors; it is being manufactured (scaled up to Kg) and used in over 25 countries. It is also in clinical trials for ovarian, pancreatic, lung, stomach and uterine cancers.[16]
But beyond the entrance of novel antibodies into clinical practice and the availability of high-throughput technologies for gene sequencing and protein characterization, the mere accumulation of knowledge in fundamental immunology has changed the landscape of the interaction between tumor and immune system, creating a new basis for development of therapeutic vaccines. Particularly important are:
- evidence of programmed contraction of the immune response;
- understanding the hallmarks of immunosenescence;
- the dual role of inflammation in the immune response;
- identification of myeloid-derived suppressor cells;
- initial characterization of cancer stem cells
- the phenomenon of immunogenic apoptosis; and
- the phenomenon of oncogene addiction.
Programmed contraction of the immune response Conventional wisdom in immunology assumed that an immune response is launched by contact with an antigen in an inflammatory context and continues until the invading antigen is cleared. Reality turned out to be more complicated. Badovinac’s 2002 paper describing experiments with mice infected by L. monocytogenes clearly showed that, after a peak of CD8 T-lymphocyte expansion at day seven, a contraction phase is initiated with kinetics independent of infection magnitude and duration.[17] That is, immune response contraction is programmed, and does not follow antigen clearance.
Since then, many new experiments have verified programmed contraction in other antigen systems, and have identified the molecules that mediate this negative feedback response.[18] The hundreds of redundant inhibitory loops or pathways are known today as immune checkpoints.[19] They are important for maintenance of self-tolerance and for protecting normal tissues from immune damage when the system reacts to infection.
The hypothesis that emerged from these data held that blockade of immune checkpoints with an antibody could amplify antitumor immunity, and it proved correct. Cytotoxic-T-lymphocyte antigen 4 (CTLA-4) is one of these immune checkpoints. Expressed in activated effector T lymphocytes, it dampens immune response by down-modulation of helper lymphocyte activity and enhancement of the immunosuppressive activity of regulatory T lymphocytes. In 2010, it was reported that a monoclonal antibody (ipilimumab) against CTLA-4 showed antitumor activity in human melanoma.[14]
Another immune checkpoint, called programmed cell death-1 (PD1) was also described as limiting T-cell activity in peripheral tissues. Additionally, persistent expression of PD1 is involved in exhaustion of the T-cell response that follows chronic antigen exposure.[18–20]
Cancer cells co-opt some of these negative feedback mechanisms to escape immune attack. Most melanoma, ovarian and lung cancer cells show high expression of the PD1 ligand.[21] Monoclonal antibodies targeting PD1 and blocking this negative feedback loop are also being tested in clinical trials for cancer treatment.[22]
Another negative feedback loop mediating immune response contraction is built on interleukin-2 (IL-2). IL-2 promotes proliferation of helper lymphocytes in the activation phase of an immune response. At the same time, it promotes expansion of regulatory T lymphocytes, which may turn off the immune response.[23] This interaction of IL2 with different cell types is mediated by its binding to different chains of the IL-2 receptor (IL2R).
At the Molecular Immunology Center (CIM, the Spanish acronym) in Havana, we have developed mathematical models to explore these complex dynamic and dual roles of IL-2.[24, 25] Moreover, we have developed mutant variants of IL-2 that bind preferentially to effector T cells or to regulatory T cells.[26] As expected, agonistic IL-2 muteins unable to bind regulatory T cells can stimulate effector cells without starting the negative feedback loop, whereas antagonistic IL-2 muteins can prevent regulatory T-cell expansion—by binding preferentially to them—allowing preferential expansion of effector cells. Both muteins show antitumor activity in experimental models.
The emerging concept is that for cancer immunotherapy to be effective, it is not enough to provoke an immune response with antibodies or vaccines. Clinical efficacy also requires some sort of therapeutic intervention on the molecular feedback loops that regulate response size and duration. Molecular targets for this intervention have been identified already.
The hallmarks of immunosenescence Cancer incidence increases with age. More than 50% of all new cases occur in people aged ≥65 years. Therefore, cancer immunotherapy must mobilize the response of a senescent immune system.
Science in this field has already characterized age-related
changes in the structure and performance of the immune system, with studies of the immune system in older adults starting to appear in the literature by the end of the 20th century. The picture that emerged is that aging is accompanied by contraction of the pool of naïve T lymphocytes, expansion of the pool of memory T lymphocytes, and by appearance of greater numbers of lymphocytes showing markers of terminal differentiation. There is also B-cell lymphopenia. Additionally, chronic inflammation is part of aging, with increasing circulating levels of pro-inflammatory cytokines such as IL-1, IL-6 and tumor necrosis factor (TNF).[27,28]
There are two underlying processes driving immunosenescence: one is reduced output of naïve lymphocytes due to involution of the thymus, and the other is chronic antigenic load that pushes available lymphocytes towards terminal differentiation. Since this process is related to contact with pathogens and antigens in the environment (and also to genetic and hormonal factors), data about the dynamics of immunosenescence obtained in one country cannot be extrapolated to another.
We have characterized the immunosenescence process in healthy Cubans of different ages, as well as in patients with lung cancer, before and after chemotherapy. García verified the aging-related decrease in number and proportion of B lymphocytes, as well as the increase in proportion of terminally differentiated CD8+ T lymphocytes, inside both CD4 helper and CD8 cytotoxic subsets.[29] Gender differences in immunosenescence were also apparent, with conservation of naïve lymphocyte output until more advanced ages in women, and higher impact of antigen-driven T-lymphocyte differentiation in men. In cancer patients, a decrease was found in the CD4/CD8 index, and chemotherapy induced an increase in IL-6 serum concentration.(unpublished CIM data).
The dual role of inflammation In 1994, the danger theory was proposed to explain tolerance to self, based on knowledge of antigen presentation and the role of dendritic antigen-presenting cells. According to danger theory, an efficient immune response requires that the antigen be presented to T lymphocytes by mature dendritic cells (otherwise the contact is tolerogenic), and maturation of dendritic cells is driven by an inflammatory environment.[30] So eliciting inflammation was considered a requirement for a cancer vaccine, precisely what adjuvants are expected to do.
But new knowledge accumulated in the next decade showed, once again, that things are not that simple, and that in some conditions inflammation can promote tumor growth—either by recruiting myeloid suppressor cells, promoting angiogenesis, or releasing reactive oxygen species, which in turn promote DNA damage and genetic instability.[31,32]
Moreover, intrinsic expression of oncogenes by cancer cells can induce proinflammatory cytokines, creating a self-reinforcing loop. Current experiments at CIM, using a model of neoplastic transformation of mesenchymal stem cells in vitro, reveal that malignant transformation initiates an inflammatory program, mediated by IL-1b and prostaglandin E2 (PGE2), which determines sustained tumorigenicity and evasion of cancer immune surveillance.[33] These experiments confirm that malignant transformation has the intrinsic potential to induce inflammation and immune evasion, even independently of any extrinsic inflammatory agent.
Complicating the situation further, this balance between cancer-controlling and cancer-promoting inflammation occurs in a senescent immune system characterized by chronic inflammation. We now suspect that the interplay among immunity, inflammation and senescence is also the basis of several aging-related chronic diseases, such as cancer, atherosclerosis, Alzheimer, osteoporosis and sarcopenia.
As we shall see later, dual roles such as the one described here for inflammation are abundant in immunology, as a consequence of the system’s intrinsic complexity.
Myeloid-derived suppressor cells One of cancer immunology’s challenges is to understand why therapeutic cancer vaccines have shown limited efficacy in clinical trials, despite existence of tumor antigens and specific T cells targeting these antigens. Tumor-induced immunosuppression is most probably the explanation: different cell populations with regulatory functions are mobilized by tumors, and contribute to restraining the immune response. Among these, recent research has identified myeloid-derived suppressor cells (MDSC) that can suppress antitumor immunity through inhibition of CD4+ and CD8+ T-lymphocyte and natural killer-cell functions.[34] MDSCs constitute a heterogeneous myelomonocytic population lacking the markers of more mature myeloid cells and expressing cell markers such as CD11b and CD33. They are generated in the bone marrow in response to cancer-derived factors such as IL-1b, TNF, and vascular endothelial growth factor, and are later recruited to the tumor site by chemokines. In cancer patients, circulating MDSC levels correlate with worse survival.[35]
The search for agents able to inhibit MDSC action is a promising avenue of work in cancer immunotherapy. At CIM, such research has concentrated on very small size proteoliposomes (VSSP), a novel adjuvant consisting of natural outer membrane vesicles derived from N. meningitides with the GM3 ganglioside hydrophobically incorporated.[36,37] Splenic MDSCs isolated from tumor-bearing mice treated with VSSP are much less immunosuppressive than tumor-induced MDSCs. Moreover, treatment with VSSP promotes differentiation of MDSCs into active antigen-presenting cells. Used as an adjuvant in therapeutic cancer vaccines, VSSP could improve antitumor efficacy by reducing tumor-induced immunosuppression, while at the same time improving antigen presentation and, consequently, T-cell activation. VSSP is currently being investigated at CIM in therapeutic clinical trials in patients with renal carcinoma, breast cancer, prostate cancer and cervical intraepithelial neoplasia grade III.
Cancer stem cells The term cancer stem cells (CSC) refers to a subset of tumor cells with the ability to self-renew and to generate a diverse set of cells inside the tumor. The cancer stem cell hypothesis posits the existence of a tumor-cell hierarchy, suggesting that tumorigenicity resides in only a small subpopulation of cancer stem cells. Early evidence for the existence of CSC came in the 1990s from studies in acute myelogenous leukemia. Later, cell subpopulations with CSC properties have been isolated from solid tumors as well. The ability to remain outside the cell cycle most of the time, together with an increased capacity for DNA repair, could make these cells more resistant to chemotherapy and radiotherapy, and thus able to repopulate the tumor and produce clinical relapse. Identification of molecular markers for CSC could lead to treatments that specifically target these cells. CD133, a molecule also called prominin-1, is one of these candidate markers for CSC, especially in brain tumors (CD133+ cells, in contrast to their CD133 counterparts, have shown ability to self-renew and undergo multilineage differentiation).[38]
At CIM, the monoclonal antibody nimotuzumab has shown antitumor activity in a brain tumor model consisting of human glioblastoma cells xenografted in athymic mice. The antibody was able to reduce the total number of radioresistant CD133+ cancer stem cells. This experiment provided the rationale for a combination of nimotuzumab and radiotherapy for glioblastoma treatment, which has already shown efficacy in clinical trials.[39]
Immunogenic apoptosis This is another emerging concept potentially key for cancer immunotherapy. It emerged in 2009, based on the observation that tumor-specific immune responses can influence outcomes of treatment with conventional cytotoxic drugs.[40,41] Until then, the supposition was that cell death through apoptosis (not necrosis) elicited immune tolerance. Now we know that under certain conditions, dying cells can be immunogenic. Moreover, we suspect that for some conventional antitumor drugs, the mechanism of antitumor action is not just directly killing cancer cells, but also depends on immunity induced by dying tumor cells. This could explain why maximal clinical response to some agents can take several months.[41]
The hallmark of immunogenic apoptosis is exposure, on the cell surface, of molecules belonging to the endoplasmic reticulum, such as the protein calreticulin. These act as a kind of “eat-me” signal for phagocytic cells, which in turn present antigens to the immune system.
At CIM, Garrido explored mechanisms of antitumor activity of antibodies against EGFR, and found that antibody blockade of EGFR was able to induce apoptosis with immunogenic characteristics. As a result of anti-EGFR treatment, apoptotic cells induced dendritic cell activation, cytotoxic lymphocytes and antitumor effect in vivo (small-molecule EGFR inhibitors did not have this property). This was the first published report of specific cytotoxic lymphocyte antitumor response generated by antibody-mediated EGFR inhibition.[42] This finding helps explain why the antitumor effect of an anti-EGFR antibody was dependent on the presence of CD4 and CD8 lymphocytes mediating active immunity. Therefore, cancer immunotherapy with anti-EGFR antibodies is not just passive receptor blockade; the antibody also has a vaccine effect, a concept with potential implications for design of therapeutic combinations.
Therapeutic induction of immunogenic apoptosis could provide an auspicious opportunity to circumvent the obstacle of high cancer-cell mutation rates. It could elicit an immune response to cancer antigens different than the one initially targeted; a phenomenon known as antigen-spreading, and which has been correlated with clinical response. At CIM, we have observed this phenomenon in melanoma patients vaccinated with an immunogenic N-glycolyl monosialoganglioside preparation who developed secondary vitiligo,[43] despite the fact that this ganglioside is not present in normal melanocytes. Antigen-spreading driven by immunogenic apoptosis opens up the possibility of inducing an immune response that evolves in parallel with the evolution of the tumor itself.
Oncogene addiction This concept was enunciated in 2002, derived from experiments showing that cancer cells are often functionally dependent on continued activity of over-expressed oncogenes to maintain the malignant phenotype.[44] Normal cells do not show such dependence. Oncogene addiction provides a rationale for therapies targeting the oncogene involved. Although cancer cells acquire mutations in multiple genes and carry several epigenetic abnormalities, they can be very sensitive to inhibition of a single oncogene.
There could be several reasons for this behavior. One is that proteins encoded by addictive oncogenes have multiple roles as they interact with metabolic or cell control circuitries. Additionally, cancer cells could be highly dependent on a single oncogene because, during the evolution of the tumor, these cells have accumulated other mutations impairing the functions of other genes that are redundant with the addictive oncogene, and therefore become less adaptable than normal cells to inhibition of that oncogene.
CIM researchers have explored the effect of monoclonal antibodies blocking EGFR from the perspective of oncogene addiction.[45] EGF addiction is apparent in patients showing over-expression of the receptor and clinical response to the humanized antibody nimotuzumab. Selection of patients carrying EGF-addicted tumors is critical for maximizing the clinical benefit of this immunotherapy. More recently, oncogene addiction has been linked to the phenomena of immunogenic apoptosis and cancer stem cells.[46] EGF binding to its receptor induces cell survival signals and up-regulation of antiapoptotic factors.[47] Functional inhibition of EGFR may trigger immunogenic cell death, which activates a T-cell mediated anti-tumor immune response.
Moreover, addiction to a given oncogene may be different in cancer stem cells than in their cellular progeny. It has been shown that combination of anti-EGFR antibodies with radiotherapy reduces the number of CD133+ cancer stem cells in glioblastoma.[39] This suggests that radiotherapy reinforces the EGFR oncogene addiction of brain cancer stem cells, adding a rationale for this therapeutic combination.
BARRIERS TO CANCER CONTROL
How to translate this accumulated scientific knowledge into new therapies? There are four formidable barriers to overcome: System complexity, tumor heterogeneity, tumor mutation rates, and human genome–environment mismatch.
“Complexity” is not just a word meaning “complicated.”[48] It is a concept emerging from mathematics: in a system of interacting components, when there are many components and nonlinear interactions, system behavior has emergent properties that cannot be predicted or attributed to any component in isolation.[49]
Redundancy and robustness to external perturbations are properties of complex systems, which are in turn barriers to the effectiveness of therapeutic interventions. This reality challenges the classical pharmacology paradigm of “one target—one drug.” Molecular interaction loops that control cellular physiology are highly redundant and therefore often result in resistance to single-target therapeutic interventions. Moreover, the outcome of blocking one molecule is context dependent (that is, dependent on the status of other connected molecules and pathways), and an intervention could be inhibitory in one circumstance and stimulatory in others; a phenomenon called dual role, which is quite common in the immune system. The dual role of IL-2 is an example, as it can stimulate effector T-lymphocyte proliferation, but can also induce overexpression of molecules mediating cell death and expand the population of regulatory T lymphocytes. It is not surprising then that attempts to treat cancer patients with high-dose IL-2 did not fulfill the promise of therapeutic efficacy.
Also complicating the picture is the fact that the outcome of blocking a molecular pathway can be exquisitely dependent on the intervention’s kinetics, reflected clinically in the phenomenon of schedule dependence: major changes in treatment response depending on dose, dose escalation and intervals.
The topology of molecular interaction networks varies from patient to patient, and from cell to cell, because of tumor cell mutation rates. Oncologists developed the concepts of heterogeneity and mutation rates many years ago through clinical observation. The novelty now is that modern technologies for DNA-sequencing can actually measure these phenomena. In normal cells, the genome replicates with high fidelity; the point mutation rate is < 10-9 per site, per cell division. Due to various mechanisms, cancer cells can mutate at 1000 times that rate, creating genome instability.[50] A scientist at the Cancer Genome Atlas Research Network characterized 178 squamous lung tumors and found a mean of 360 exonic mutations per tumor and recurrent mutations in 11 genes.[51]
As techniques for DNA sequencing improved, inter-s and intra-tumor genetic heterogeneity was found to be greater than expected. Very few mutations were repeated in more than 5%–10% of tumors of a particular tissue type.[52]
Complexity, heterogeneity and mutation rates explain why cell culture experiments and even animal models used to test therapeutic interventions fail so often to predict treatment outcomes in clinical settings. The reductionist approach of experimental research implies the creation of experimental models, in which the molecular interactions to be studied are isolated from other “confounding” variables and are protagonists for the biological behavior of that model. Experimental results then fail to translate into clinical situations in which the targeted molecule acts in the context of multiple redundant regulatory loops, coded by genes that mutate rapidly and differently from one patient to another. These are formidable barriers.
RECOGNIZING EVOLUTIONARY CONSTRAINTS
Some barriers to cancer immunotherapy are set by evolution itself. The immune system exists as it is, with all its complexity, because it has been selected by evolution to guarantee fitness and reproductive performance in a given environment. In biology, causation takes the form of adaptive selection. But the human immune system evolved in a high-mortality environment that is not the same as the one in which we live today.[53]
Most mammals have a life expectancy just slightly longer than their reproductive life and die from extrinsic causes acting early in life, such as infections, starvation or predators. In contrast, modern humans usually live decades beyond their reproductive life, with low mortality kinetics until aging, and die mostly from intrinsic age-related diseases or senescence. But this is a development of the last 200 years. Homo sapiens evolved over 150,000 years in a high-mortality environment, with low life expectancy and early death from extrinsic causes, such as infections. Thus, the immune system has been tuned to provide survival advantage until reproductive age in precisely these conditions. The very same traits (such as strong inflammatory reactions) that provide survival advantage against infections in youth can be deleterious beyond reproductive age, by promoting inflammation-related chronic diseases. This contradiction has been called antagonistic pleiotropy.[54]
Phenomena such as programmed immune response contraction and inflammatory senescence make a lot of sense from this evolutionary perspective. We now live longer, in a low-mortality environment, and our immune system has not been selected for providing advantage in this context, creating a genome–environment mismatch. Natural selection, driven by reproductive advantage, does not operate beyond reproductive age, which is precisely when cancer incidence is highest. It is not surprising then that the immune system has fewer inbuilt mechanisms to control cancer than it has to control infections.
THE WAY FORWARD: IMPLICATIONS FOR CANCER RESEARCH
In this contradictory landscape of flourishing scientific production and barriers to its use, what scientific strategies can be suggested? I propose six:
- address cancer as a chronic disease;
- find relevant biomarkers for patient stratification;
- create a rationale for therapeutic combinations;
- target immune response control loops;
- expand mathematical modeling capacity; and
- evaluate complex intervention packages in real-world conditions.
Cancer, even in its advanced forms, is becoming a chronic disease. Cuban studies of survival in disseminated stages of cancer found 5-year survival of 30% for breast, 32% for uterine corpus, 24% for oral cavity and 20% for colon cancers.[55]
Modern targeted drugs, directed at the products of mutated or overexpressed specific genes, and biotechnology drugs—including monoclonal antibodies and cancer vaccines—generally show lower toxicity profiles than conventional chemotherapy, and are therefore compatible with long-term use. These drugs can foster the trend towards chronicity.
Survival curves using data from Cuba’s National Cancer Registry and from clinical trials of lung cancer therapeutic vaccines at CIM suggest the existence of two distinct patient subpopulations, one of them with a more chronic evolution.
The transition from a rapidly fatal disease to a chronic condition is not new in the history of medicine. It is exactly what happened with diabetes mellitus after the discovery of insulin in 1921 and the series of technological improvements that ensued. Analogous histories could be described for cardiovascular diseases and kidney failure.
The concept of chronicity has major implications for clinical research. The current dominant paradigm in cancer clinical trials arose from an extrapolation of experience with classical cytotoxic drugs. According to it, antitumor activity is connected to toxicity, and treatments should be scaled up to the maximum tolerated dose; pharmacokinetics is important to define optimal dosage; an active drug must produce rapid tumor shrinkage; and this short-term response is a predictor of long-term survival.
Immunotherapy—which acts indirectly on the tumor to improve survival and quality of life in an intrinsically chronic disease—needs a different paradigm, according to which dosage is not directly related to pharmacokinetics, survival improvement can be obtained without tumor shrinkage, and the therapeutic effect could be delayed in time and continue even after disease progression. Clinical trials should take this into account.
Patient stratification into subgroups defined by relevant biological markers is another component of the strategy. We are now in an era of molecular subtypes.[56] An early example more than 30 years ago was the adoption of routine determination of estrogen and progesterone receptors as predictors of short-term breast cancer relapse.[57] But the biotechnology revolution has multiplied the possibilities to measure molecular markers, both in nucleic acids (high throughput sequencing, gene expression) and in proteins (proteomics), and the issue now is to select which measurements are relevant for a given treatment.
Some examples: After hormone-receptor measurements became widely available, Her2-expression determination was introduced as a predictor of breast cancer response to the monoclonal antibody trastuzumab.[58] The identification of mutations in EGFR and ALK genes enabled prediction of response to novel tyrosine-kinase inhibitors in lung cancer.[59,60] Overexpression of EGFR in tumor samples is related to probability of response to the monoclonal antibody nimotuzumab.[61] And increased serum EGF concentrations identify lung cancer patients who will benefit from the therapeutic EGF vaccine.[62]
The search for appropriate markers should not be limited to tumor molecular characterization, but be extended to study of systemic response, (particularly markers of immunosenescence), potentially relevant for immunotherapy.
The complexity of regulatory molecular networks driving cancer-cell proliferation and their interactions with the immune system make it unlikely we can modify the biology of the system by acting only on one node of the network. Targeting gene products involved in oncogene addiction may be the exception, but as a rule, the barrier of complexity should be addressed with therapeutic combinations.
The problem today is not a lack of possible combinations, but the potential existence of too many. With just one chemotherapy regime, plus a therapeutic vaccine and a couple of monoclonal antibodies, there are 24 possible combinations and permutations of the three components; the number would escalate quickly if testing diverse dosages and schedules.
This vast expanse of possibilities is impossible to explore by trial and error in clinical settings. The challenge is to develop a theoretical framework for immunotherapy combinations in order to reduce the space to be explored. Basic immunology is still far from putting forward such a theory, but there is already enough emerging knowledge to propose, by way of a preliminary hypothesis, some organizing principles to be validated in experiments and clinical settings:
- reduce tumor burden as much as possible by conventional means;
- introduce therapeutic vaccines after chemotherapy, preferring to vaccinate in lymphopenia;
- combine therapeutic vaccines with cytotoxic drugs known to induce immunogenic apoptosis;
- direct monoclonal antibodies and vaccines at targets known to be related to oncogene addiction;
- combine therapeutic vaccines and antibodies with intervention on immune system regulatory components;
- continue maintenance immunotherapy long term, even after disease progression;
- treat chronic systemic inflammation if present; and
- test therapeutic combinations for tumor stabilization and survival improvements, not for short-term response rate.
Biological drugs interfering with negative feedback loops that control the immune response and mediate its contraction would be critical components of these combinations. To date, the literature has described monoclonal antibodies targeting CTLA-4 and PD1, two molecules involved in negative regulation of immune response.[19]
At CIM, we are exploring muteins of IL-2[26] designed by computer simulation to show reduced affinity for the IL-2 receptor alpha chain (hence reduced effect on T-regulatory lymphocytes) or reduced affinity for the receptor’s gamma chain (preferentially blocking expansion of T-regulatory lymphocytes). The antitumor action of these muteins and their roles in therapeutic combinations are being investigated.
Among the toughest challenges of complexity are schedule dependence and dual roles. Interferon gamma, for example, induces Th1-helper lymphocyte activity, but can recruit myeloid suppressor cells and induce PD1L expression in cancer cells. IL-2 promotes proliferation of helper T lymphocytes, but can also promote regulatory T-cell expansion, which may shut off the immune response. The dynamic balance between these opposite effects will depend on doses and treatment timing, and cannot be predicted by intuition.
CIM system biology staff have developed mathematical models for helper, regulatory and memory T-cell dynamics that can now be used to simulate the effect of various types of therapy to potentiate either immunity or tolerance.[63] As we deepen our understanding of the complexity of tumor/immunity interactions, we will need more mathematical models to assist our intuition in designing therapeutic combinations and schedules to be tested in preclinical or clinical experiments. Mathematical models can point to counterintuitive conclusions, such as the feasibility of vaccinating in lymphopenia or the advantage of regimes that maintain tumor stabilization over those looking for tumor shrinkage.[64] Such predictions must, of course, be validated by experiments.
Finally, addressing complexity in immunotherapy requires more scientific research conducted in real-world conditions. The theory of controlled clinical trials reflects an attempt to perform clinical research in conditions as similar as possible to those of experimental research: evaluate new treatments one at a time, in homogeneous populations of selected patients, and isolate the effect of treatment to remove any confounding effects of concomitant interventions. This approach has been fertile in providing new drugs backed by hard data, but it has also created distortions. One of them is reduced representation of the real patient population. The proportion of real cancer patients enrolled in clinical trials is as low as 2–3%, and older patients are usually underrepresented. Interactions of new cancer treatments with other comorbidities—and their treatments—are not usually seen or examined in clinical trials. These are some of the reasons why many “advances” in cancer treatment revealed in clinical trials have not translated into real impact in oncology practice.
Management of chronic non-communicable diseases requires complex and long interventions including diverse components, none of which may produce high impact in isolation. They should be tested using a scientific methodology, but as part of a package that includes not only oncospecific treatments, but also palliative care, nutritional support, pain control and other components. And they should be evaluated in unselected patient populations, in real-world conditions, mainly in primary care settings. Certainly, this approach is not a substitute for rigorous clinical trials, but it is a much-needed complement.
CONCLUSION: RECOGNIZING THE PARADIGM SHIFT
The idea of harnessing the immune system to fight cancer has made its way into clinical practice, and the tools to do it (cytokines, monoclonal antibodies, therapeutic vaccines and probably also cell therapy) are being provided by biotechnology. But this undeniable advance came simultaneously with the understanding of the complexity of the basic biological mechanisms involved. Cancer physiopathology itself is rooted in these mechanisms, through which life emerged and works, constituting a disruption of the biological order that occurs through many possible molecular pathways.
Science is always an exercise of reductionism: we isolate causes and effects as much as possible, and look for the simplest explanation for what we observe. Albert Einstein said, “The grand aim of all science is to cover the greatest number of empirical facts by logical deduction from the smallest number of hypotheses and axioms.”[65] In medical sciences, such a reductionist strategy has been helpful in fighting infectious diseases, given the possibility of identifying a single microbial cause and rather simple interventions to attack it. However, this approach does not work that well for chronic diseases, which have multiple causes and threaten health through disruption of diverse mechanisms of normal physiology.
That is why the simultaneous entry of immunotherapy and complexity into cancer research comes with a sense of paradigm shift. Health systems will need to handle an expanding population of active persons living with cancer. For many patients the goal will not be cure, but long-term control. Pathologists will classify tumors in molecular terms. Clinical research will move from testing drugs, to testing complex intervention packages. Scientists will increasingly use mathematical simulation to generate hypotheses before performing experiments. The scientific method will permeate medical practice even in primary care settings. And borders between knowledge creation in experimental conditions and knowledge validation in real-life conditions will become blurred, drawing a continuous gradient of complexity and predictive capacity from fundamental research to population-wide health interventions. We must be prepared for all of this.