
Immune Response to SARS-CoV-2 Infection in Obesity and T2D: Literature Review

 , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
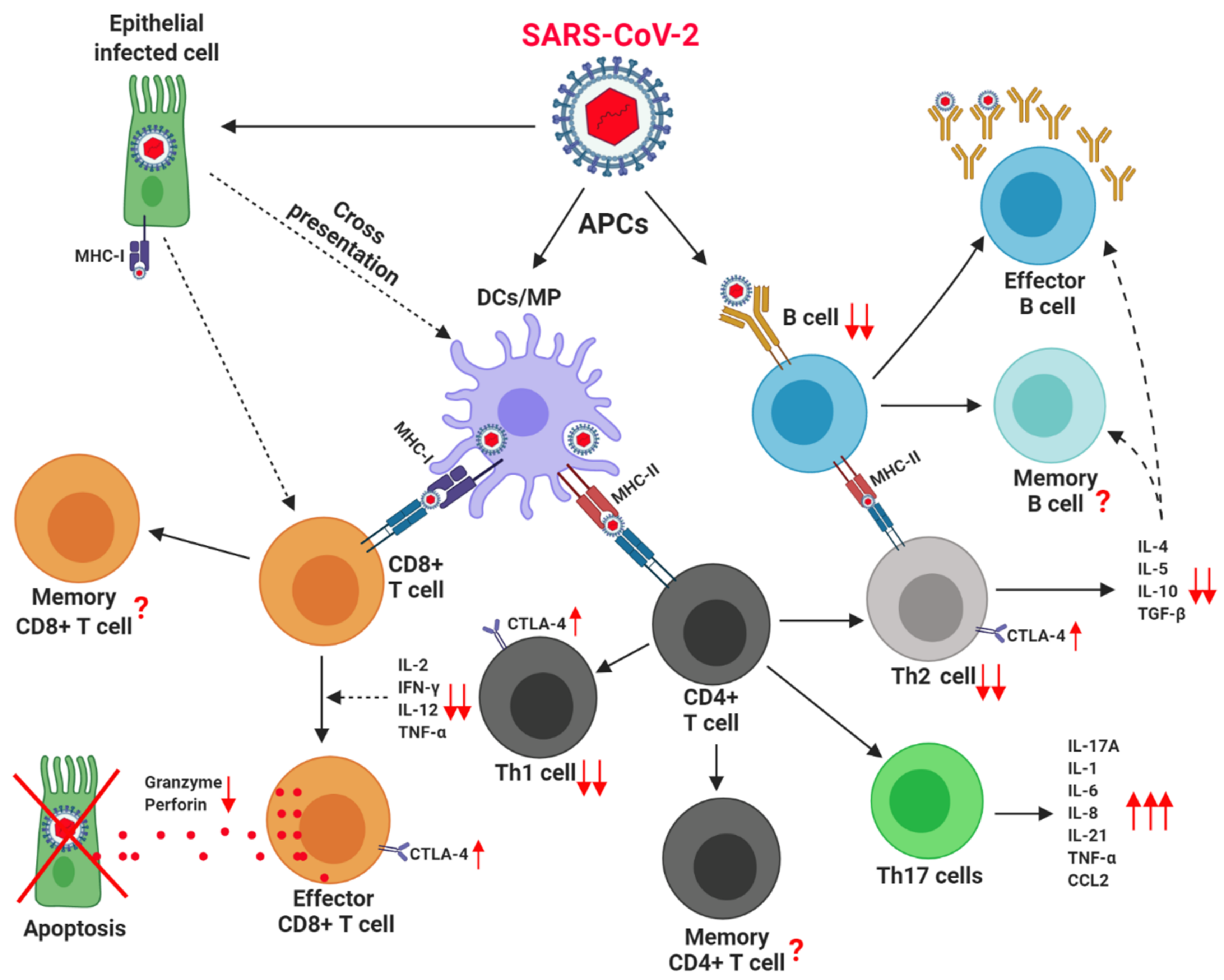
2. How Immune System Fight against Virus
2.1. Role of Macrophages and Dendritic Cells against Virus
2.2. Role of CD8+ T Cells against Virus
2.3. Role of CD4+ T Cells against Virus
2.4. Role of B Cells against Virus
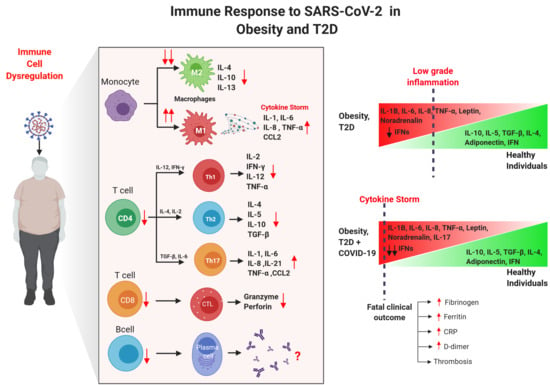
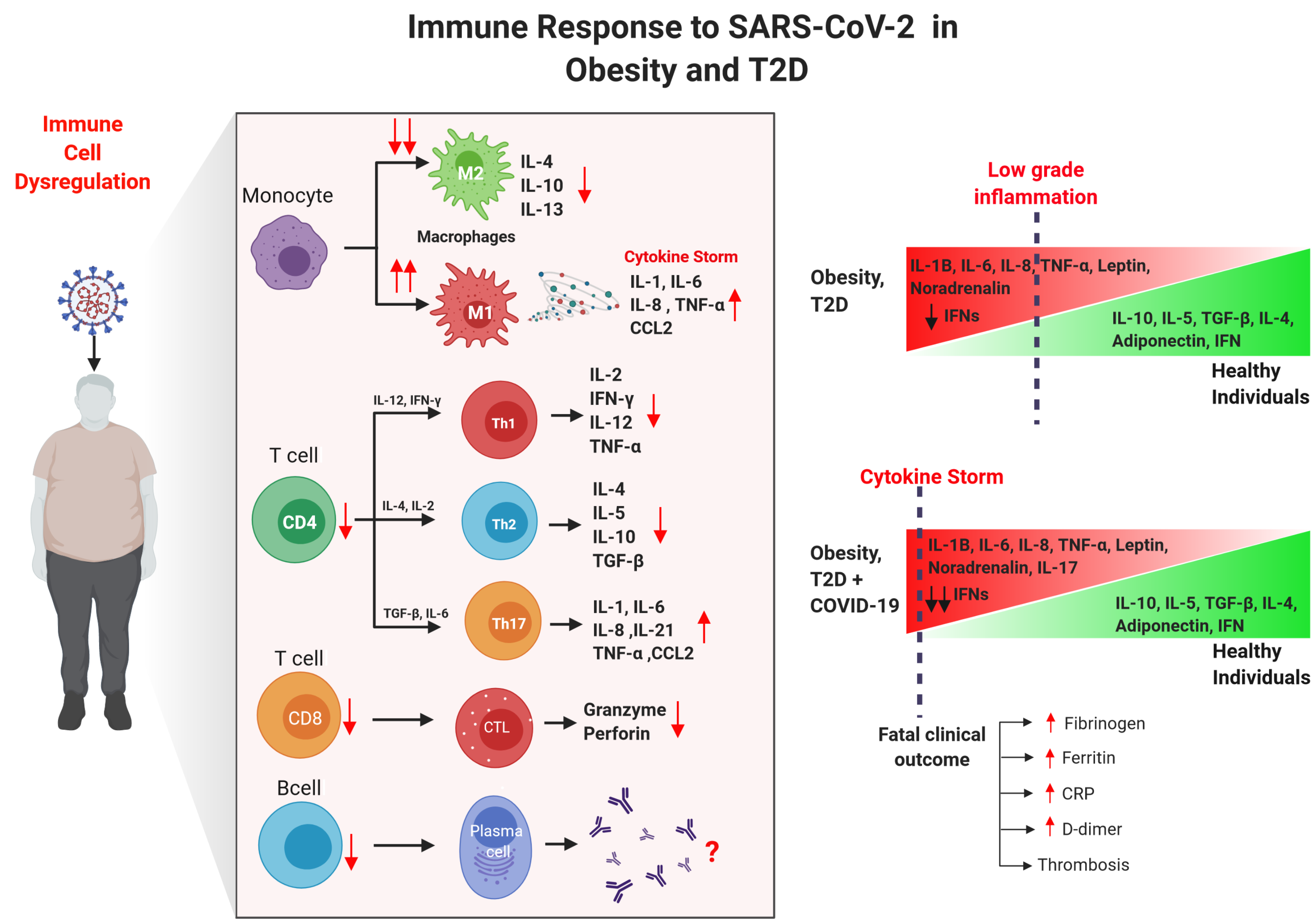
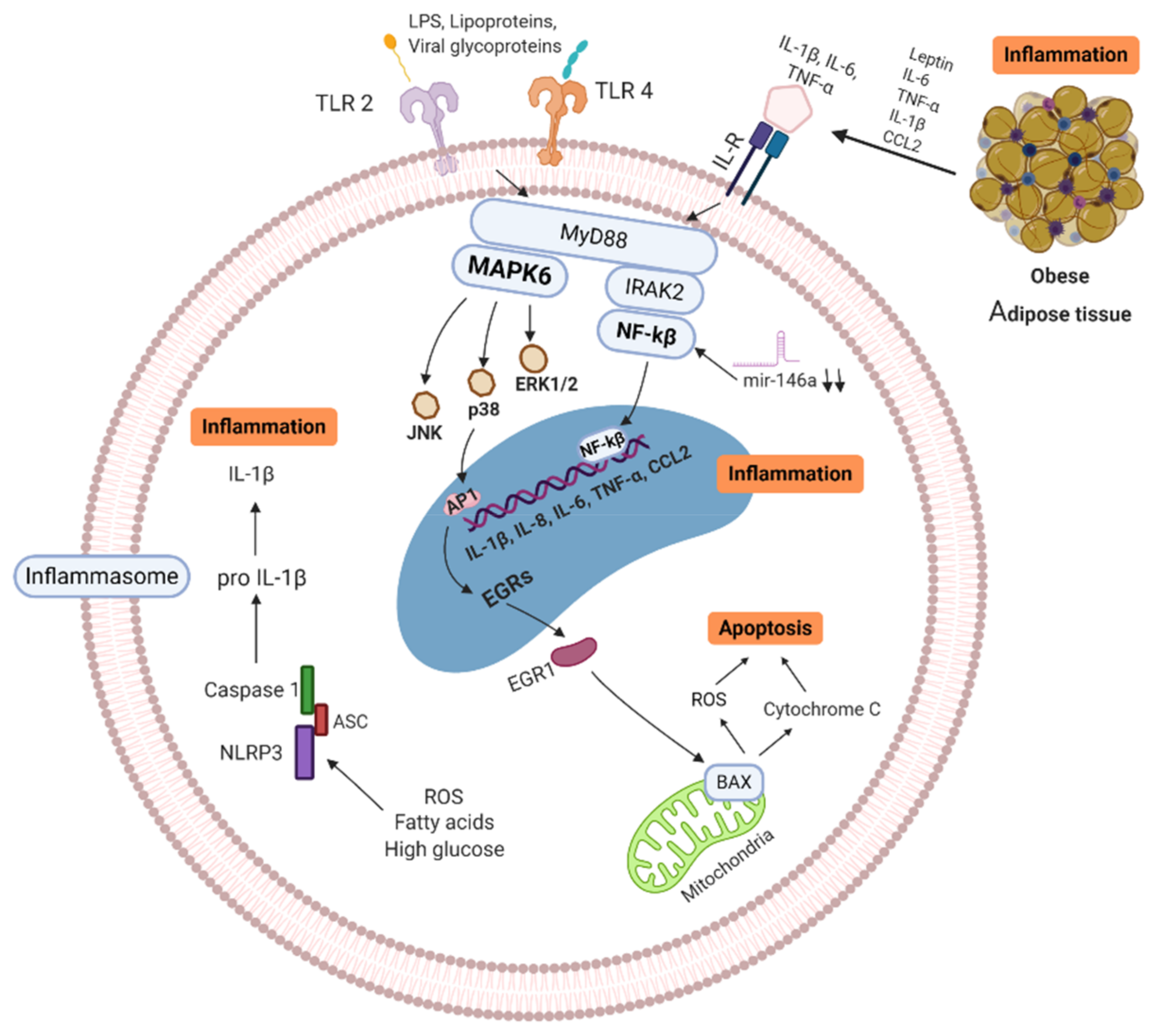
3. Immune Alterations in Obesity and T2D
4. Impact of Obesity and T2D in COVID-19 Patients
4.1. Clinical Characteristics of Obese/T2D Patients with COVID-19
4.2. Immune Innate Responses of Obese/T2D Patients with COVID-19
4.3. Immune Adaptive Responses of Obese/T2D Patients with COVID-19
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chehrehgosha, M. The Unpreparedness of the Healthcare System for the Management of COVID-19 Pandemic Leading to the Mistreatment of the Elderly: A Newly Emerging Moral Dilemma. J. Nutr. Heal. Aging 2020. [Google Scholar] [CrossRef]
- Gallo Marin, B.; Aghagoli, G.; Lavine, K.; Yang, L.; Siff, E.J.; Chiang, S.S.; Salazar-Mather, T.P.; Dumenco, L.; Savaria, M.C.; Aung, S.N.; et al. Predictors of COVID-19 severity: A literature review. Rev. Med. Virol. 2020, e2146. [Google Scholar] [CrossRef]
- Nieman, D.C. Coronavirus disease-2019: A tocsin to our aging, unfit, corpulent, and immunodeficient society. J. Sport Health Sci. 2020. [Google Scholar] [CrossRef]
- Cucinotta, D.; Vanelli, M. WHO declares COVID-19 a pandemic. Acta Biomed. 2020. [Google Scholar] [CrossRef]
- Johns Hopkins University. COVID-19 Map—Johns Hopkins Coronavirus Resource Center; Johns Hopkins Coronavirus Resource Center: Baltimore, MA, USA, 2020. [Google Scholar]
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S.; Rajagopal, S.; Pai, A.R.; Kutty, S. Cytokine Storm in COVID-19—Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol. 2020, 11, 1648. [Google Scholar] [CrossRef]
- Deng, Y.; Liu, W.; Liu, K.; Fang, Y.Y.; Shang, J.; Zhou, L.; Wang, K.; Leng, F.; Wei, S.; Chen, L.; et al. Clinical characteristics of fatal and recovered cases of coronavirus disease 2019 in Wuhan, China: A retrospective study. Chin. Med. J. 2020, 133, 1261–1267. [Google Scholar] [CrossRef]
- Chiappetta, S.; Sharma, A.M.; Bottino, V.; Stier, C. COVID-19 and the role of chronic inflammation in patients with obesity. Int. J. Obes. 2020. [Google Scholar] [CrossRef]
- Yan, Y.; Yang, Y.; Wang, F.; Ren, H.; Zhang, S.; Shi, X.; Yu, X.; Dong, K. Clinical characteristics and outcomes of patients with severe covid-19 with diabetes. BMJ Open Diabetes Res. Care 2020. [Google Scholar] [CrossRef]
- Vaamonde, J.G.; Álvarez-Món, M.A. Obesity and overweight. Medicine 2020. [Google Scholar] [CrossRef]
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; IDF: Brussels, Belgium, 2019; ISBN 9782930229874. [Google Scholar]
- Muniyappa, R.; Gubbi, S. COVID-19 pandemic, coronaviruses, and diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 2020. [Google Scholar] [CrossRef] [Green Version]
- Ryan, P.M.D.; Caplice, N.M. Is Adipose Tissue a Reservoir for Viral Spread, Immune Activation, and Cytokine Amplification in Coronavirus Disease 2019? Obesity 2020, 28, 1191–1194. [Google Scholar] [CrossRef] [Green Version]
- Daryabor, G.; Atashzar, M.R.; Kabelitz, D.; Meri, S.; Kalantar, K. The Effects of Type 2 Diabetes Mellitus on Organ Metabolism and the Immune System. Front. Immunol. 2020. [Google Scholar] [CrossRef]
- de Lucena, T.M.C.; da Silva Santos, A.F.; de Lima, B.R.; de Albuquerque Borborema, M.E.; de Azevêdo Silva, J. Mechanism of inflammatory response in associated comorbidities in COVID-19. Diabetes Metab. Syndr. Clin. Res. Rev. 2020. [Google Scholar] [CrossRef]
- Dai, S.P.; Zhao, X.; Wu, J.H. Effects of Comorbidities on the Elderly Patients with COVID-19: Clinical Characteristics of Elderly Patients Infected with COVID-19 from Sichuan, China. J. Nutr. Heal. Aging 2020. [Google Scholar] [CrossRef]
- Cruz-Tapias, P.; Castiblanco, J.; Correa, N.E.; Montoya-Ortíz, G. AUTOIMMUNITY From Bench to Bedside; El Rosario University Press: Bogota, Colombia, 2013; ISBN 978-958-738-376-8. [Google Scholar]
- Janeway, C.A. How the immune system protects the host from infection. Microbes Infect. 2001. [Google Scholar] [CrossRef]
- Frieman, M.; Heise, M.; Baric, R. SARS coronavirus and innate immunity. Virus Res. 2008. [Google Scholar] [CrossRef]
- Lee, H.K.; Iwasaki, A. Innate control of adaptive immunity: Dendritic cells and beyond. Semin. Immunol. 2007. [Google Scholar] [CrossRef]
- Mathan, T.S.M.; Figdor, C.G.; Buschow, S.I. Human plasmacytoid dendritic cells: From molecules to intercellular communication network. Front. Immunol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Kotsias, F.; Cebrian, I.; Alloatti, A. Antigen processing and presentation. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780128183519. [Google Scholar]
- Zuniga, E.I.; McGavern, D.B.; Oldstone, M.B.A. Antigen Presentation. In Encyclopedia of Virology; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Castellino, F.; Germain, R.N. Cooperation between CD4+ and CD8+ T cells: When, where, and how. Annu. Rev. Immunol. 2006. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K.; Libby, P.; Schönbeck, U.; Yan, Z.Q. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ. Res. 2002. [Google Scholar] [CrossRef]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010. [Google Scholar] [CrossRef]
- Garcia, K.C.; Teyton, L.; Wilson, I.A. Structural basis of T cell recognition. Annu. Rev. Immunol. 1999. [Google Scholar] [CrossRef]
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988. [Google Scholar] [CrossRef]
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The structural basis of antibody-antigen recognition. Front. Immunol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Lanzavecchia, A.; Sallusto, F. Antigen decoding by T lymphocytes: From synapses to fate determination. Nat. Immunol. 2001. [Google Scholar] [CrossRef]
- Mempel, T.R.; Henrickson, S.E.; Von Andrian, U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Walunas, T.L.; Bluestone, J.A. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 1996. [Google Scholar] [CrossRef]
- Condotta, S.A.; Richer, M.J. The immune battlefield: The impact of inflammatory cytokines on CD8+T-cell immunity. PLoS Pathog. 2017, 13, e1006618. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T ceils to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef]
- Van Kooten, G.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17. [Google Scholar] [CrossRef]
- Ara, A.; Ahmed, K.A.; Xiang, J. Multiple effects of CD40–CD40L axis in immunity against infection and cancer. ImmunoTargets Ther. 2018. [Google Scholar] [CrossRef] [Green Version]
- Xiang, J.; Huang, H.; Liu, Y. A New Dynamic Model of CD8 + T Effector Cell Responses via CD4 + T Helper-Antigen-Presenting Cells. J. Immunol. 2005. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Yao, S.; Chen, L. Cell Surface Signaling Molecules in the Control of Immune Responses: A Tide Model. Immunity 2011. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.A.; Xiang, J. Mechanisms of cellular communication through intercellular protein transfer. J. Cell. Mol. Med. 2011. [Google Scholar] [CrossRef]
- Quezada, S.A.; Jarvinen, L.Z.; Lind, E.F.; Noelle, R.J. CD40/CD154 interactions at the interface of tolerance and immunity. Annu. Rev. Immunol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bevan, M.J. CD8+ T Cells: Foot Soldiers of the Immune System. Immunity 2011, 35, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harty, J.T.; Tvinnereim, A.R.; White, D.W. Cd8+ T cell effector mechanisms in resistance to infection. Annu. Rev. Immunol. 2000. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, C.; Rocha, B.; Tanchot, C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science 2002. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.; Cantrell, D.A. Metabolism, migration and memory in cytotoxic T cells. Nat. Rev. Immunol. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schluns, K.S.; Lefrançois, L. Cytokine control of memory T-cell development and survival. Nat. Rev. Immunol. 2003. [Google Scholar] [CrossRef]
- Embgenbroich, M.; Burgdorf, S. Current concepts of antigen cross-presentation. Front. Immunol. 2018. [Google Scholar] [CrossRef] [Green Version]
- van Montfoort, N.; van der Aa, E.; Woltman, A.M. Understanding MHC class I presentation of viral antigens by human dendritic cells as a basis for rational design of therapeutic vaccines. Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012. [Google Scholar] [CrossRef]
- Schönbeck, U.; Libby, P. The CD40/CD154 receptor/ligand dyad. Cell. Mol. Life Sci. 2001. [Google Scholar] [CrossRef]
- Sant, A.J.; McMichael, A. Revealing the role of CD4+ T cells in viral immunity. J. Exp. Med. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, N.R.; Chevalier, M.S.; Johnson, D.C. Viral inhibition of MHC class II antigen presentation. Trends Immunol. 2003. [Google Scholar] [CrossRef]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding roles for CD4 + T cells in immunity to viruses. Nat. Rev. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Machado, P.R.L.; Carvalho, L.; Araújo, M.I.A.S.; Carvalho, E.M. Immune response mechanisms to infections. An. Bras. Dermatol. 2004. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, T.; Takeda, K.; Tanaka, T.; Ohkusu, K.; Kashiwamura, S.; Okamura, H.; Akira, S.; Nakanishi, K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: Synergism with IL-18 for IFN-gamma production. J. Immunol. 1998, 161, 3400–3407. [Google Scholar] [PubMed]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 2001. [Google Scholar] [CrossRef] [PubMed]
- Appay, V. The physiological role of cytotoxic CD4+ T-cells: The holy grail? Clin. Exp. Immunol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Casazza, J.P.; Betts, M.R.; Price, D.A.; Precopio, M.L.; Ruff, L.E.; Brenchley, J.M.; Hill, B.J.; Roederer, M.; Douek, D.C.; Koup, R.A. Acquisition of direct antiviral effector functions by CMV-specific CD4 + T lymphocytes with cellular maturation. J. Exp. Med. 2006. [Google Scholar] [CrossRef]
- Soghoian, D.Z.; Jessen, H.; Flanders, M.; Sierra-Davidson, K.; Cutler, S.; Pertel, T.; Ranasinghe, S.; Lindqvist, M.; Davis, I.; Lane, K.; et al. HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef] [Green Version]
- Baumgarth, N. How specific is too specific? B-cell responses to viral infections reveal the importance of breadth over depth. Immunol. Rev. 2013. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, S.F.; Lukacs-Kornek, V.; Kuligowski, M.P.; Pitcher, L.A.; Degn, S.E.; Kim, Y.A.; Cloninger, M.J.; Martinez-Pomares, L.; Gordon, S.; Turley, S.J.; et al. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat. Immunol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.H.; Baumgarth, N. The Multifaceted B Cell Response to Influenza Virus. J. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotty, S. A brief history of T cell help to B cells. Nat. Rev. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Rothaeusler, K.; Baumgarth, N. B-cell fate decisions following influenza virus infection. Eur. J. Immunol. 2010. [Google Scholar] [CrossRef] [Green Version]
- Basso, K.; Klein, U.; Niu, H.; Stolovitzky, G.A.; Tu, Y.; Califano, A.; Cattoretti, G.; Dalla-Favera, R. Tracking CD40 signaling during germinal center development. Blood 2004. [Google Scholar] [CrossRef]
- MacLennan, I.C.M. Germinal centers. Annu. Rev. Immunol. 1994. [Google Scholar] [CrossRef]
- Scheid, J.F.; Mouquet, H.; Feldhahn, N.; Seaman, M.S.; Velinzon, K.; Pietzsch, J.; Ott, R.G.; Anthony, R.M.; Zebroski, H.; Hurley, A.; et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 2009. [Google Scholar] [CrossRef]
- Yu, X.; Tsibane, T.; McGraw, P.A.; House, F.S.; Keefer, C.J.; Hicar, M.D.; Tumpey, T.M.; Pappas, C.; Perrone, L.A.; Martinez, O.; et al. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature 2008. [Google Scholar] [CrossRef] [Green Version]
- Dörner, T.; Radbruch, A. Antibodies and B Cell Memory in Viral Immunity. Immunity 2007. [Google Scholar] [CrossRef] [Green Version]
- Hooijkaas, H.; Benner, R.; Pleasants, J.R.; Wostmann, B.S. Isotypes and specificities of immunoglobulins produced by germ-free mice fed chemically defined ultrafiltered “antigen-free” diet. Eur. J. Immunol. 1984. [Google Scholar] [CrossRef]
- Goossens, G.H.; Dicker, D.; Farpour-Lambert, N.J.; Fruhbeck, G.; Mullerova, D.; Woodward, E.; Holm, J.C. Obesity and COVID-19: A Perspective from the European Association for the Study of Obesity on Immunological Perturbations, Therapeutic Challenges, and Opportunities in Obesity. Obes. Facts 2020, 13, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Luo, S.; Gui, Y.; Zhou, H.; Zhang, Z.; Tian, C.; Zhou, Q.; Wang, Q.; Hu, Y.; Fan, H.; et al. Obesity is a potential risk factor contributing to clinical manifestations of COVID-19. Int. J. Obes. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ritter, A.; Kreis, N.N.; Louwen, F.; Yuan, J. Obesity and covid-19: Molecular mechanisms linking both pandemics. Int. J. Mol. Sci. 2020, 21, 5793. [Google Scholar] [CrossRef] [PubMed]
- Méry, G.; Epaulard, O.; Borel, A.L.; Toussaint, B.; Le Gouellec, A. COVID-19: Underlying Adipokine Storm and Angiotensin 1-7 Umbrella. Front. Immunol. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, S.D.; Fiedler, J.; Bauersachs, J.; Thum, T.; Sedding, D.G. Senescence-induced inflammation: An important player and key therapeutic target in atherosclerosis. Eur. Heart J. 2020. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.-L.; Tsai, W.-C.; Hung, R.-W.; Chen, I.-Y.; Shu, K.-H.; Pan, S.-Y.; Yang, F.-J.; Ting, T.-T.; Jiang, J.-Y.; Peng, Y.-S.; et al. Emergence of T cell immunosenescence in diabetic chronic kidney disease. Immun. Ageing 2020. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016. [Google Scholar] [CrossRef]
- Beumer, W.; Drexhage, R.C.; De Wit, H.; Versnel, M.A.; Drexhage, H.A.; Cohen, D. Increased level of serum cytokines, chemokines and adipokines in patients with schizophrenia is associated with disease and metabolic syndrome. Psychoneuroendocrinology 2012. [Google Scholar] [CrossRef] [Green Version]
- Ortega, E.; Gálvez, I.; Martín-Cordero, L. Adrenergic Regulation of Macrophage-Mediated Innate/Inflammatory Responses in Obesity and Exercise in this Condition: Role of β2 Adrenergic Receptors. Endocr. Metab. Immune Disord. Drug Targets 2019. [Google Scholar] [CrossRef]
- Martín-Cordero, L.; Gálvez, I.; Hinchado, M.D.; Ortega, E. Influence of Obesity and Exercise on β2-Adrenergic-Mediated Anti-Inflammatory Effects in Peritoneal Murine Macrophages. Biomedicines 2020, 8, 556. [Google Scholar] [CrossRef]
- Martín-Cordero, L.; García, J.J.; Hinchado, M.D.; Ortega, E. The interleukin-6 and noradrenaline mediated inflammation-stress feedback mechanism is dysregulated in metabolic syndrome: Effect of exercise. Cardiovasc. Diabetol. 2011. [Google Scholar] [CrossRef] [Green Version]
- García, J.J.; del Carmen Sáez, M.; De la Fuente, M.; Ortega, E. Regulation of phagocytic process of macrophages by noradrenaline and its end metabolite 4-hydroxy-3-metoxyphenyl-glycol. Role of α- and β-adrenoreceptors. Mol. Cell. Biochem. 2003. [Google Scholar] [CrossRef]
- Martín-Cordero, L.; Gálvez, I.; Hinchado, M.D.; Ortega, E. β2 Adrenergic Regulation of the Phagocytic and Microbicide Capacity of Macrophages from Obese and Lean Mice: Effects of Exercise. Nutrients 2019, 11, 2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef] [Green Version]
- Aly, O.; Zaki, H.H.; Herzalla, M.R.; Fathy, A.; Raafat, N.; Hafez, M.M. Gene polymorphisms of Patatin-like phospholipase domain containing 3 (PNPLA3), adiponectin, leptin in diabetic obese patients. PLoS ONE 2020, 15, e0234465. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Maehata, E.; Yano, M.; Taniyama, M.; Suzuki, S. Correlation between the adiponectin-leptin ratio and parameters of insulin resistance in patients with type 2 diabetes. Metabolism 2005, 54, 281–286. [Google Scholar] [CrossRef]
- Bidulescu, A.; Dinh, P.C.; Sarwary, S.; Forsyth, E.; Luetke, M.C.; King, D.B.; Liu, J.; Davis, S.K.; Correa, A. Associations of leptin and adiponectin with incident type 2 diabetes and interactions among African Americans: The Jackson heart study. BMC Endocr. Disord. 2020, 20. [Google Scholar] [CrossRef] [Green Version]
- Thorand, B.; Zierer, A.; Baumert, J.; Meisinger, C.; Herder, C.; Koenig, W. Associations between leptin and the leptin/adiponectin ratio and incident Type 2 diabetes in middle-aged men and women: Results from the MONICA/KORA Augsburg Study 1984–2002. Diabet. Med. 2010, 27, 1004–1011. [Google Scholar] [CrossRef]
- Van Dielen, F.M.H.; Van’t Veer, C.; Schols, A.M.; Soeters, P.B.; Buurman, W.A.; Greve, J.W.M. Increased leptin concentrations correlate with increased concentrations of inflammatory markers in morbidly obese individuals. Int. J. Obes. 2001, 25, 1759–1766. [Google Scholar] [CrossRef] [Green Version]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, fat mass and immune system: Role for leptin. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Satoh, N.; Naruse, M.; Usui, T.; Tagami, T.; Suganami, T.; Yamada, K.; Kuzuya, H.; Shimatsu, A.; Ogawa, Y. Leptin-to-adiponectin ratio as a potential atherogenic index in obese type 2 diabetic patients. Diabetes Care 2004, 27, 2488–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hamodi, Z.; Al-Habori, M.; Al-Meeri, A.; Saif-Ali, R. Association of adipokines, leptin/adiponectin ratio and C-reactive protein with obesity and type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, N.; Imamura, S.; Fujita, T.; Uchida, Y.; Inagaki, K.; Kakizawa, H.; Hayakawa, N.; Suzuki, A.; Takeda, J.; Horikawa, Y.; et al. The ratio of leptin to adiponectin can be used as an index of insulin resistance. Metabolism 2008, 57, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Finucane, F.M.; Luan, J.; Wareham, N.J.; Sharp, S.J.; O’Rahilly, S.; Balkau, B.; Flyvbjerg, A.; Walker, M.; Højlund, K.; Nolan, J.J.; et al. Correlation of the leptin: Adiponectin ratio with measures of insulin resistance in non-diabetic individuals. Diabetologia 2009, 52, 2345–2349. [Google Scholar] [CrossRef] [Green Version]
- Kotani, K.; Sakane, N.; Saiga, K.; Kurozawa, Y. Leptin: Adiponectin ratio as an atherosclerotic index in patients with type 2 diabetes: Relationship of the index to carotid intima-media thickness. Diabetologia 2005, 48, 2684–2686. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, M.; Maruoka, S.; Katayose, S. Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur. J. Endocrinol. 2002, 147, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Kolfschoten, I.G.M.; Roggli, E.; Nesca, V.; Regazzi, R. Role and therapeutic potential of microRNAs in diabetes. Diabetes, Obes. Metab. 2009, 11, 118–129. [Google Scholar] [CrossRef]
- Landrier, J.F.; Derghal, A.; Mounien, L. MicroRNAs in Obesity and Related Metabolic Disorders. Cells 2019, 8, 859. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.A.; Ludwig, R.G.; Garcia-Martin, R.; Brandão, B.B.; Kahn, C.R. Extracellular miRNAs: From Biomarkers to Mediators of Physiology and Disease. Cell Metab. 2019. [Google Scholar] [CrossRef]
- DP, B. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function.pdf. Cell 2004, 116, 281–297. [Google Scholar]
- McClelland, A.D.; Kantharidis, P. microRNA in the development of diabetic complications. Clin. Sci. 2014, 126, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; Enlund, E.; Funcke, J.B.; Tews, D.; Holzmann, K.; Debatin, K.M.; Wabitsch, M.; Fischer-Posovszky, P. MiR-146a-mediated suppression of the inflammatory response in human adipocytes. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Q.; Brichard, S.; Yi, X.; Li, Q. MicroRNAs as a new mechanism regulating adipose tissue inflammation in obesity and as a novel therapeutic strategy in the metabolic syndrome. J. Immunol. Res. 2014, 2014, 987285. [Google Scholar] [CrossRef] [Green Version]
- Tili, E.; Michaille, J.-J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b Levels following Lipopolysaccharide/TNF-α Stimulation and Their Possible Roles in Regulating the Response to Endotoxin Shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Ståhle, M.; Pivarcsi, A. MicroRNAs and immunity: Novel players in the regulation of normal immune function and inflammation. Semin. Cancer Biol. 2008, 18, 131–140. [Google Scholar] [CrossRef]
- Baldeón R, L.; Weigelt, K.; de Wit, H.; Ozcan, B.; van Oudenaren, A.; Sempértegui, F.; Sijbrands, E.; Grosse, L.; Freire, W.; Drexhage, H.A.; et al. Decreased serum level of miR-146a as sign of chronic inflammation in type 2 diabetic patients. PLoS ONE 2014, 9, e115209. [Google Scholar] [CrossRef] [Green Version]
- Delhanty, P.J.D.; Huisman, M.; Baldeon-rojas, L.Y.; Van Den Berge, I.; Grefhorst, A.; Abribat, T.; Leenen, P.J.M.; Themmen, A.P.N.; Van Der Lely, A. Des-acyl ghrelin analogs prevent high-fat-diet-induced dysregulation of glucose homeostasis. FASEB J. 2013, 27, 1690–1700. [Google Scholar] [CrossRef]
- Xia, C.; Rao, X.; Zhong, J. Role of T Lymphocytes in Type 2 Diabetes and Diabetes-Associated Inflammation. J. Diabetes Res. 2017. [Google Scholar] [CrossRef]
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009. [Google Scholar] [CrossRef]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Eller, K.; Kirsch, A.; Wolf, A.M.; Sopper, S.; Tagwerker, A.; Stanzl, U.; Wolf, D.; Patsch, W.; Rosenkranz, A.R.; Eller, P. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Chaudhry, A.; Kas, A.; DeRoos, P.; Kim, J.M.; Chu, T.T.; Corcoran, L.; Treuting, P.; Klein, U.; Rudensky, A.Y. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control TH2 responses. Nature 2009. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Mraz, M.; Haluzik, M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J. Endocrinol. 2014, 222, R113–R127. [Google Scholar] [CrossRef] [Green Version]
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011. [Google Scholar] [CrossRef]
- Simar, D.; Versteyhe, S.; Donkin, I.; Liu, J.; Hesson, L.; Nylander, V.; Fossum, A.; Barrès, R. DNA methylation is altered in B and NK lymphocytes in obese and type 2 diabetic human. Metabolism 2014. [Google Scholar] [CrossRef]
- Ip, B.; Cilfone, N.A.; Belkina, A.C.; Defuria, J.; Jagannathan-Bogdan, M.; Zhu, M.; Kuchibhatla, R.; McDonnell, M.E.; Xiao, Q.; Kepler, T.B.; et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFα production. Obesity 2016. [Google Scholar] [CrossRef]
- Shade, K.-T.; Anthony, R. Antibody Glycosylation and Inflammation. Antibodies 2013, 2, 392–414. [Google Scholar] [CrossRef] [Green Version]
- Alter, G.; Ottenhoff, T.H.M.; Joosten, S.A. Antibody glycosylation in inflammation, disease and vaccination. Semin. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Li, H.; Liu, D.; Tao, L.; Zhang, J.; Liang, B.; Liu, X.; Wang, X.; Li, X.; Wang, Y.; et al. IgG Glycosylation Profile and the Glycan Score Are Associated with Type 2 Diabetes in Independent Chinese Populations: A Case-Control Study. J. Diabetes Res. 2020. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, A.; Favre, G.; Frey, S.; Esnault, V.; Gugenheim, J.; Bouam, S.; Schiavo, L.; Tran, A.; Alifano, M. Obesity and COVID-19: ACE 2, the Missing Tile. Obes. Surg. 2020. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.J.; Liu, J.; Chen, Y.; Ye, B.; Li, N.; Wang, X.; Tang, M.; Shao, J. Characteristics of laboratory findings of COVID-19 patients with comorbid diabetes mellitus. Diabetes Res. Clin. Pract. 2020. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, Z.; Liu, X.; Liu, G.; Xie, D.; Xu, Z.; Zhao, J.; Zhang, J. Clinical features and potential risk factors for discerning the critical cases and predicting the outcome of patients with COVID-19. J. Clin. Lab. Anal. 2020. [Google Scholar] [CrossRef]
- Cai, S.H.; Liao, W.; Chen, S.W.; Liu, L.L.; Liu, S.Y.; Zheng, Z.D. Association between obesity and clinical prognosis in patients infected with SARS-CoV-2. Infect. Dis. Poverty 2020, 9, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wei, Y.; Chen, M.; Wan, Q.; Chen, X. Clinical analysis of risk factors for severe COVID-19 patients with type 2 diabetes. J. Diabetes Complicat. 2020, 34, 107666. [Google Scholar] [CrossRef] [PubMed]
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M.; et al. High Prevalence of Obesity in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) Requiring Invasive Mechanical Ventilation. Obesity 2020. [Google Scholar] [CrossRef]
- Qin, W.; Hu, B.Z.; Zhang, Z.; Chen, S.; Li, F.J.; Zhu, Z.Y.; Wang, X.J.; Liu, M.; Li, C.H. Clinical characteristics and death risk factors of severe COVID-19. Zhonghua Jie He He Hu Xi Za Zhi 2020. [Google Scholar] [CrossRef]
- Kass, D.A. COVID-19 and Severe Obesity: A Big Problem? Ann. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Zhao, R.; Sun, Y.; Zhang, Y.; Wang, W.; Wang, S.; Wang, C.; Liu, J.; Gao, L.; Hu, Z.; Fei, J.; et al. Distinguishable Immunologic Characteristics of COVID-19 Patients with Comorbid Type 2 Diabetes Compared with Nondiabetic Individuals. Mediat. Inflamm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gabarre, P.; Dumas, G.; Dupont, T.; Darmon, M.; Azoulay, E.; Zafrani, L. Acute kidney injury in critically ill patients with COVID-19. Intensive Care Med. 2020. [Google Scholar] [CrossRef]
- Becker, R.C. COVID-19-associated vasculitis and vasculopathy. J. Thromb. Thrombolysis 2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Geng, X.; Tan, Y.; Li, Q.; Xu, C.; Xu, J.; Hao, L.; Zeng, Z.; Luo, X.; Liu, F.; et al. New understanding of the damage of SARS-CoV-2 infection outside the respiratory system. Biomed. Pharmacother. 2020. [Google Scholar] [CrossRef]
- Lee, S.G.; Fralick, M.; Sholzberg, M. Coagulopathy associated with COVID-19. CMAJ 2020. [Google Scholar] [CrossRef]
- Quah, P.; Li, A.; Phua, J.; Phua, J. Mortality rates of patients with COVID-19 in the intensive care unit: A systematic review of the emerging literature. Crit. Care 2020. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Mahawar, K.; Xia, Z.; Yang, W.; EL-Hasani, S. Obesity and mortality of COVID-19. Meta-analysis. Obes. Res. Clin. Pract. 2020. [Google Scholar] [CrossRef]
- Morgan, O.W.; Bramley, A.; Fowlkes, A.; Freedman, D.S.; Taylor, T.H.; Gargiullo, P.; Belay, B.; Jain, S.; Cox, C.; Kamimoto, L.; et al. Morbid obesity as a risk factor for hospitalization and death due to 2009 pandemic influenza A(H1N1) disease. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Umpierrez, G.E.; Kovatchev, B.P. Glycemic Variability: How to Measure and Its Clinical Implication for Type 2 Diabetes. Am. J. Med. Sci. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kota, S.; Modi, K.; Satya Krishna, S. Glycemic variability: Clinical implications. Indian J. Endocrinol. Metab. 2013. [Google Scholar] [CrossRef]
- Hulme, K.D.; Gallo, L.A.; Short, K.R. Influenza virus and glycemic variability in diabetes: A killer combination? Front. Microbiol. 2017. [Google Scholar] [CrossRef]
- Marshall, R.J.; Armart, P.; Hulme, K.D.; Chew, K.Y.; Brown, A.C.; Hansbro, P.M.; Bloxham, C.J.; Flint, M.; Ronacher, K.; Bielefeldt-Ohmann, H.; et al. Glycemic variability in diabetes increases the severity of influenza. MBio 2020. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, Y.; Arima, H.; Zoungas, S.; Ninomiya, T.; Cooper, M.; Hamet, P.; Mancia, G.; Poulter, N.; Harrap, S.; Woodward, M.; et al. Impact of visit-to-visit glycemic variability on the risks of macrovascular and microvascular events and all-cause mortality in type 2 diabetes: The ADVANCE trial. Diabetes Care 2014. [Google Scholar] [CrossRef] [Green Version]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Martinelli, L.; Motz, E.; Ceriello, A. Intermittent High Glucose Enhances Apoptosis Related to Oxidative Stress in Human Umbilical Vein Endothelial Cells: The Role of Protein Kinase C and NAD(P)H-Oxidase Activation. Diabetes 2003. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Wang, N.; Wang, J.; Luo, A.; Gao, F.; Tu, Y. Admission fasting plasma glucose is an independent risk factor for 28-day mortality in patients with COVID-19. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Milionis, C.; Milioni, S.O. A brief analysis and hypotheses about the risk of COVID-19 for people with type 1 and type 2 diabetes mellitus. J. Diabetes Metab. Disord. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wei, W.; Yang, K.; Li, S.; Yu, X.; Dong, C.; Zhang, B. Glycemic control before admission is an important determinant of prognosis in patients with coronavirus disease 2019. J. Diabetes Investig. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, S.; Rankawat, G.; Singh, A.; Gupta, V.; Kakkar, S. Impact of glycemic control in diabetes mellitus on management of COVID-19 infection. Int. J. Diabetes Dev. Ctries. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, H.; Pollock, C.; Ma, J.; Wu, H.; Chadban, S.; Panchapakesan, U. The role of TLR2 and 4-mediated inflammatory pathways in endothelial cells exposed to high glucose. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chi, J.; Lv, W.; Wang, Y. Obesity and diabetes as high-risk factors for severe coronavirus disease 2019 (Covid-19). Diabetes. Metab. Res. Rev. 2020. [Google Scholar] [CrossRef]
- Roncon, L.; Zuin, M.; Rigatelli, G.; Zuliani, G. Diabetic patients with COVID-19 infection are at higher risk of ICU admission and poor short-term outcome. J. Clin. Virol. 2020. [Google Scholar] [CrossRef]
- Pal, R.; Banerjee, M. Are people with uncontrolled diabetes mellitus at high risk of reinfections with COVID-19? Prim. Care Diabetes 2020. [Google Scholar] [CrossRef]
- Mishra, Y.; Pathak, B.K.; Mohakuda, S.S.; Tilak, T.V.S.V.G.K.; Sen, S.; P, H.; Singh, R.; Singh, A.R. Relation of D-dimer levels of COVID-19 patients with diabetes mellitus. Diabetes Metab. Syndr. Clin. Res. Rev. 2020. [Google Scholar] [CrossRef]
- Zhu, L.; She, Z.G.; Cheng, X.; Qin, J.J.; Zhang, X.J.; Cai, J.; Lei, F.; Wang, H.; Xie, J.; Wang, W.; et al. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-existing Type 2 Diabetes. Cell Metab. 2020. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Xiang, P.; Pu, L.; Xiong, H.; Li, C.; Zhang, M.; Tan, J.; Xu, Y.; Song, R.; et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J. Transl. Med. 2020, 18, 206. [Google Scholar] [CrossRef]
- Urra, J.M.; Cabrera, C.M.; Porras, L.; Ródenas, I. Selective CD8 cell reduction by SARS-CoV-2 is associated with a worse prognosis and systemic inflammation in COVID-19 patients. Clin. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tatum, D.; Taghavi, S.; Houghton, A.; Stover, J.; Toraih, E.; Duchesne, J. Neutrophil-to-Lymphocyte Ratio and Outcomes in Louisiana COVID-19 Patients. Shock 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, X.; Chen, J.; Jin, Y.; Peng, L.; Wang, H.H.X.; Luo, M.; Chen, L.; Zhao, Y. Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Huang, W.; Ye, B.; Chen, C.; Huang, R.; Wu, F.; Wei, Q.; Zhang, W.; Hu, J. Changes of hematological and immunological parameters in COVID-19 patients. Int. J. Hematol. 2020. [Google Scholar] [CrossRef]
- Coppelli, A.; Giannarelli, R.; Aragona, M.; Penno, G.; Falcone, M.; Tiseo, G.; Ghiadoni, L.; Barbieri, G.; Monzani, F.; Virdis, A.; et al. Hyperglycemia at hospital admission is associated with severity of the prognosis in patients hospitalized for COVID-19: The pisa COVID-19 study. Diabetes Care 2020. [Google Scholar] [CrossRef]
- Qu, R.; Ling, Y.; Zhang, Y.H.Z.; Wei, L.Y.; Chen, X.; Li, X.M.; Liu, X.Y.; Liu, H.M.; Guo, Z.; Ren, H.; et al. Platelet-to-lymphocyte ratio is associated with prognosis in patients with coronavirus disease-19. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Otsuka, R.; Seino, K.I. Macrophage activation syndrome and COVID-19. Inflamm. Regen. 2020. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy Eur. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Ho, Y.C. SARS-CoV-2: A storm is raging. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Mahmudpour, M.; Roozbeh, J.; Keshavarz, M.; Farrokhi, S.; Nabipour, I. COVID-19 cytokine storm: The anger of inflammation. Cytokine 2020. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F. Aging, male sex, obesity, and metabolic inflammation create the perfect storm for COVID-19. Diabetes 2020, 69, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Grom, A.A.; Mellins, E.D. Macrophage activation syndrome: Advances towards understanding pathogenesis. Curr. Opin. Rheumatol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020. [Google Scholar] [CrossRef]
- Wang, Z.; Pan, H.; Jiang, B. Type I IFN deficiency: An immunological characteristic of severe COVID-19 patients. Signal Transduct. Target. Ther. 2020. [Google Scholar] [CrossRef]
- Lee, J.S.; Shin, E.C. The type I interferon response in COVID-19: Implications for treatment. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Hirashima, T.; Arai, T.; Kitajima, H.; Tamura, Y.; Yamada, T.; Hashimoto, S.; Morishita, H.; Minamoto, S.; Kawashima, K.; Kashiwa, Y.; et al. Factors significantly associated with COVID-19 severity in symptomatic patients: A retrospective single-center study. J. Infect. Chemother. 2020, 27, 76–82. [Google Scholar] [CrossRef]
- Jurado, A.; Martín, M.C.; Abad-Molina, C.; Orduña, A.; Martínez, A.; Ocaña, E.; Yarce, O.; Navas, A.M.; Trujillo, A.; Fernández, L.; et al. COVID-19: Age, Interleukin-6, C-reactive protein, and lymphocytes as key clues from a multicentre retrospective study. Immun. Ageing 2020, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Nie, H.X.; Hu, K.; Wu, X.J.; Zhang, Y.T.; Wang, M.M.; Wang, T.; Zheng, Z.S.; Li, X.C.; Zeng, S.L. Abnormal immunity of non-survivors with COVID-19: Predictors for mortality. Infect. Dis. Poverty 2020, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Dai, L.; Zhang, Y.; Fu, W.; Gao, Y.; Zhang, Z.; Zhang, Z. Clinical Characteristics and Risk Factors for Disease Severity and Death in Patients With Coronavirus Disease 2019 in Wuhan, China. Front. Med. 2020, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Alamdari, N.M.; Afaghi, S.; Rahimi, F.S.; Tarki, F.E.; Tavana, S.; Zali, A.; Fathi, M.; Besharat, S.; Bagheri, L.; Pourmotahari, F.; et al. Mortality risk factors among hospitalized COVID-19 patients in a major referral center in Iran. Tohoku J. Exp. Med. 2020. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Y.; Li, J.; Han, H.; Xia, Z.; Liu, F.; Wu, K.; Yang, L.; Liu, X.; Zhu, C. Decreased T cell populations contribute to the increased severity of COVID-19. Clin. Chim. Acta 2020. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wang, J.; Wang, Y.; Zhao, J.; Huang, J.; Tian, Y.; Yang, C.; Zhang, H.; Zhang, M.; Gu, L.; et al. The Metabolic Changes and Immune Profiles in Patients With COVID-19. Front. Immunol. 2020. [Google Scholar] [CrossRef]
- Wang, F.; Qu, M.; Zhou, X.; Zhao, K.; Lai, C.; Tang, Q.; Xian, W.; Chen, R.; Li, X.; Li, Z.; et al. The timeline and risk factors of clinical progression of COVID-19 in Shenzhen, China. J. Transl. Med. 2020. [Google Scholar] [CrossRef]
- Chen, Z.; John Wherry, E. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Schub, D.; Klemis, V.; Schneitler, S.; Mihm, J.; Lepper, P.M.; Wilkens, H.; Bals, R.; Eichler, H.; Gärtner, B.C.; Becker, S.L.; et al. High levels of SARS-CoV-2–specific T cells with restricted functionality in severe courses of COVID-19. JCI Insight 2020. [Google Scholar] [CrossRef]
- Jouan, Y.; Guillon, A.; Gonzalez, L.; Perez, Y.; Boisseau, C.; Ehrmann, S.; Ferreira, M.; Daix, T.; Jeannet, R.; François, B.; et al. Phenotypical and functional alteration of unconventional T cells in severe COVID-19 patients. J. Exp. Med. 2020. [Google Scholar] [CrossRef]
- Long, Q.X.; Liu, B.Z.; Deng, H.J.; Wu, G.C.; Deng, K.; Chen, Y.K.; Liao, P.; Qiu, J.F.; Lin, Y.; Cai, X.F.; et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat. Med. 2020, 26, 845–848. [Google Scholar] [CrossRef] [PubMed]
- De Biasi, S.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Gozzi, L.; Iannone, A.; Lo Tartaro, D.; Mattioli, M.; et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yang, X.O. TH17 responses in cytokine storm of COVID-19: An emerging target of JAK2 inhibitor Fedratinib. J. Microbiol. Immunol. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Holloway, T.L.; Rani, M.; Cap, A.P.; Stewart, R.M.; Schwacha, M.G. The association between the Th-17 immune response and pulmonary complications in a trauma ICU population. Cytokine 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlov, M.; Wander, P.L.; Morrell, E.D.; Mikacenic, C.; Wurfel, M.M. A Case for Targeting Th17 Cells and IL-17A in SARS-CoV-2 Infections. J. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ibarrondo, F.J.; Fulcher, J.A.; Goodman-Meza, D.; Elliott, J.; Hofmann, C.; Hausner, M.A.; Ferbas, K.G.; Tobin, N.H.; Aldrovandi, G.M.; Yang, O.O. Rapid Decay of Anti–SARS-CoV-2 Antibodies in Persons with Mild Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Sang, L.; Ye, F.; Ruan, S.; Zhong, B.; Song, T.; Alshukairi, A.N.; Chen, R.; Zhang, Z.; et al. Kinetics of viral load and antibody response in relation to COVID-19 severity. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Bölke, E.; Matuschek, C.; Fischer, J.C. Loss of Anti–SARS-CoV-2 Antibodies in Mild Covid-19. N. Engl. J. Med. 2020, 383, 1697–1698. [Google Scholar] [CrossRef]
- Neidich, S.D.; Green, W.D.; Rebeles, J.; Karlsson, E.A.; Schultz-Cherry, S.; Noah, T.L.; Chakladar, S.; Hudgens, M.G.; Weir, S.S.; Beck, M.A. Increased risk of influenza among vaccinated adults who are obese. Int. J. Obes. 2017. [Google Scholar] [CrossRef] [Green Version]
- Green, W.D.; Beck, M.A. Obesity impairs the adaptive immune response to influenza virus. Ann. Am. Thorac. Soc. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hemalatha Rajkumar, P.B. The Impact of Obesity on Immune Response to Infection and Vaccine: An Insight into Plausible Mechanisms. Endocrinol. Metab. Syndr. 2013. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Osornio, S.A.; Cruz-Hernández, T.R.; Drago-Serrano, M.E.; Campos-Rodríguez, R. Immunity to influenza: Impact of obesity. Obes. Res. Clin. Pract. 2019. [Google Scholar] [CrossRef] [PubMed]
- Michiels, B.; Govaerts, F.; Remmen, R.; Vermeire, E.; Coenen, S. A systematic review of the evidence on the effectiveness and risks of inactivated influenza vaccines in different target groups. Vaccine 2011. [Google Scholar] [CrossRef] [PubMed]
- Paich, H.A.; Sheridan, P.A.; Handy, J.; Karlsson, E.A.; Schultz-Cherry, S.; Hudgens, M.G.; Noah, T.L.; Weir, S.S.; Beck, M.A. Overweight and obese adult humans have a defective cellular immune response to pandemic H1N1 Influenza a virus. Obesity 2013. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, P.A.; Paich, H.A.; Handy, J.; Karlsson, E.A.; Hudgens, M.G.; Sammon, A.B.; Holland, L.A.; Weir, S.; Noah, T.L.; Beck, M.A. Obesity is associated with impaired immune response to influenza vaccination in humans. Int. J. Obes. 2012. [Google Scholar] [CrossRef] [Green Version]
- Sell, H.; Habich, C.; Eckel, J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012. [Google Scholar] [CrossRef]
- Park, H.L.; Shim, S.H.; Lee, E.Y.; Cho, W.; Park, S.; Jeon, H.J.; Ahn, S.Y.; Kim, H.; Nam, J.H. Obesity-induced chronic inflammation is associated with the reduced efficacy of influenza vaccine. Hum. Vaccines Immunother. 2014. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, J.K.; Kim, D.J.; Nam, J.H.; Shim, S.M.; Choi, Y.K.; Lee, C.H.; Poo, H. Diet-induced obesity dramatically reduces the efficacy of a 2009 pandemic H1N1 vaccine in a mouse model. J. Infect. Dis. 2012. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, E.A.; Sheridan, P.A.; Beck, M.A. Diet-induced obesity in mice reduces the maintenance of influenza-specific CD8+ memory T cells. J. Nutr. 2010. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Galarza, J.; Prócel, C.; Cañadas, C.; Aguirre, D.; Pibaque, R.; Bedón, R.; Sempértegui, F.; Drexhage, H.; Baldeón, L. Immune Response to SARS-CoV-2 Infection in Obesity and T2D: Literature Review. Vaccines 2021, 9, 102. https://doi.org/10.3390/vaccines9020102
Pérez-Galarza J, Prócel C, Cañadas C, Aguirre D, Pibaque R, Bedón R, Sempértegui F, Drexhage H, Baldeón L. Immune Response to SARS-CoV-2 Infection in Obesity and T2D: Literature Review. Vaccines. 2021; 9(2):102. https://doi.org/10.3390/vaccines9020102
Chicago/Turabian StylePérez-Galarza, Jorge, César Prócel, Cristina Cañadas, Diana Aguirre, Ronny Pibaque, Ricardo Bedón, Fernando Sempértegui, Hemmo Drexhage, and Lucy Baldeón. 2021. "Immune Response to SARS-CoV-2 Infection in Obesity and T2D: Literature Review" Vaccines 9, no. 2: 102. https://doi.org/10.3390/vaccines9020102