
Abstract
The pandemic influenza 2009 (A(H1N1)pdm09) virus currently causes seasonal and annual epidemic outbreaks. The widespread use of anti-influenza drugs such as neuraminidase and matrix protein 2 (M2) channel inhibitors has resulted in the emergence of drug-resistant influenza viruses. In this study, we aimed to determine the anti-influenza A(H1N1)pdm09 virus activity of azithromycin, a re-positioned macrolide antibiotic with potential as a new anti-influenza candidate, and to elucidate its underlying mechanisms of action. We performed in vitro and in vivo studies to address this. Our in vitro approaches indicated that progeny virus replication was remarkably inhibited by treating viruses with azithromycin before infection; however, azithromycin administration after infection did not affect this process. We next investigated the steps inhibited by azithromycin during virus invasion. Azithromycin did not affect attachment of viruses onto the cell surface, but blocked internalization into host cells during the early phase of infection. We further demonstrated that azithromycin targeted newly budded progeny virus from the host cells and inactivated their endocytic activity. This unique inhibitory mechanism has not been observed for other anti-influenza drugs, indicating the potential activity of azithromycin before and after influenza virus infection. Considering these in vitro observations, we administered azithromycin intranasally to mice infected with A(H1N1)pdm09 virus. Single intranasal azithromycin treatment successfully reduced viral load in the lungs and relieved hypothermia, which was induced by infection. Our findings indicate the possibility that azithromycin could be an effective macrolide for the treatment of human influenza.
Similar content being viewed by others


Introduction
Influenza A viruses cause annual epidemics that peak during winter season, frequently leading to an increase in hospitalizations and deaths, mainly among the elderly and infants [1]. Historically, the occurrence of an influenza outbreak has often led to huge casualties. The Spanish influenza A(H1N1) of 1918 resulted in a worldwide pandemic that caused massive devastation, with an estimated 20–50 million deaths [2]. In 1997, a highly pathogenic avian influenza A(H5N1) virus was first recognized as capable of infecting humans; sporadic human infections with this virus have resulted in a fatality rate greater than 50% due to severe respiratory disease [3]. A novel infectious virus influenza A(H1N1)pdm09 virus triggered the most recent global pandemic decades ago [4]. Further, severe respiratory diseases were evoked by the pandemic virus in several cases [5]. Influenza-induced severe respiratory disease leads to a high fatality rate owing to respiratory disorders and failure. Extracorporeal membrane oxygenation for respiratory failure induced by A(H1N1)pdm09 virus has shown limited success in Japan (35.7% survival rate) [6]. Thus, the development and/or repositioning of anti-influenza agents that can reduce the viral load are necessary. Clinically used neuraminidase inhibitors are beneficial for human influenza; they inhibit progeny virus yield during the acute phase of infection [7, 8]. However, a marked increase in drug-resistant A(H1N1) viruses was observed, and they are currently an emerging problem worldwide [9,10,11]. Recently, new antiviral agents have been approved for the treatment of influenza in Japan. Favipiravir inactivates RNA-dependent RNA polymerases of broad-spectrum RNA viruses including influenza viruses [12]. Nevertheless, this RNA polymerase inhibitor induces some toxicities, limiting its clinical use. Xofluza™, a cap-dependent endonuclease inhibitor that has been recently approved for influenza, blocks the initiation of virus mRNA synthesis in host cells [13]; however, viruses resistant to Xofluza™ have emerged [14]. Thus, strategies to prepare for and protect against the next outbreak of influenza, as well as current seasonal influenza, are essential. The development of novel anti-influenza drugs from the basics is a time-consuming process. Therefore, repositioning different types of licensed drugs is one of the most validated strategies to identify new anti-influenza drugs within a short period.
Accordingly, one antibiotic, clarithromycin (CAM), a 14-membered macrolide, is effective against influenza virus infection. Moreover, the anti-influenza virus activities of CAM have been supported by both in vivo and in vitro studies [15, 16]. Previously, our group reported that a 16-membered macrolide, leucomycin A3 (LM-A3, also called as josamycin), shows noticeable anti-influenza A virus activities based on both in vivo and in vitro studies [17]. The synthesized 12-membered EM900 macrolide, in which anti-bacterial activity was eliminated, also resulted in a survival advantage in mice infected with influenza A(H1N1) virus [17]. These reports indicate that different membered ring structures of macrolides show diverse anti-virus activity. The 15-membered macrolides such as azithromycin (AZM) are considered promising anti-influenza agents. Prior to proceeding with our present study, we aimed to identify macrolide candidates from different membered ring structure macrolides including 12-, 14-, 15-, and 16-membered variants that exert inhibitory effects on the activities of influenza A(H1N1)pdm09 virus. We found that AZM shows anti-A(H1N1)pdm09 virus activity by in vitro screening. In this study, via in vitro approaches, we demonstrated that AZM exerts anti-influenza A(H1N1)pdm09 virus activity via a mechanism different from that associated with other currently available anti-influenza drugs including macrolides. Based on this underlying antiviral mechanism, we further elucidated that AZM can ameliorate pathological status in vivo. Our findings could broaden the treatment options for influenza epidemics and suggest an alternative strategy to develop and design anti-influenza therapeutics.
Materials and methods
Macrolide compound
Azithromycin dehydrate for all experiments was purchased from Tokyo Chemical Industry Co., Ltd (TCI, Japan).
Cells
Human A549 and MDCK (Madin-Darby canine kidney) cells were grown and maintained in supplemented Dulbecco’s modified Eagle's medium (DMEM) or minimum essential medium (MEM) (Sigma Life Science, United Kingdom) with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 μg ml−1 streptomycin, and 100 μg ml−1 penicillin.
Virus
Human influenza A(H1N1)pdm09 (A/California/7/2009 (H1N1)) virus was supplied from A-CLIP institute under the guidelines of Chiba University (Chiba, Japan). MDCK cells were infected with the viruses and cultured for 24 h. Next, virus titers in the culture medium were determined using a viral plaque assay, described subsequently.
To prepare mouse-adapted influenza A(H1N1)pdm09 virus, 8-week-old female mice were anesthetized with isoflurane, and were intranasally infected with 1 × 104 plaque forming units (pfu) of the human influenza A(H1N1)pdm09 virus. After 4 days, lung tissues were homogenized in 1.5 ml PBS. After centrifuging at 8000 r.p.m. for 5 min, the supernatant was collected and diluted three times with RPMI 1640 medium supplemented with 2% FBS. A 30-μl aliquot was used for the second inoculum, and the previously described steps were repeated 10 times. The last passaged virus was used for the following animal experiments.
Different azithromycin treatments against virus infection in host cells
AZM was dissolved in EtOH and adjusted to achieve a final concentration of 0.2% EtOH in DMEM. A confluent monolayer of A549 cells was infected with A(H1N1)pdm09 virus at a multiplicity of infection (MOI) of 1 under four different treatment conditions with 200 µM of AZM as follows (i) post-infection treatment: A549 cells were infected with the viruses at 35 °C for 1 h. After infection, the cells were washed with PBS and cultured in 2 ml supplemented DMEM with or without AZM at 37 °C for 48 h. (ii) Pretreatment of cells: the host cells were pretreated with 300 μl non-supplemented DMEM with or without AZM at 37 °C for 1 h. After removal of the medium, the cells were washed with PBS and infected with the viruses at 35 °C for 1 h. Subsequently, the cells were washed with PBS and cultured with 2 ml AZM-free supplemented DMEM at 37 °C for 48 h. (iii) Pretreatment of viruses: the viruses were pretreated with 300 μl non-supplemented DMEM with or without AZM for 1 h at 37 °C. After the treatment, A549 cells were infected with the viruses for 1 h at 35 °C. Then, the cells were washed with PBS and cultured in 2 ml AZM-free supplemented DMEM at 37 °C for 48 h. (iv) Treatment at the time of infection: viruses were premixed with 300 μl non-supplemented DMEM in the absence or presence of AZM, and A549 cells were immediately infected for 1 h at 35 °C. After infection, the cells were washed with PBS and cultured in 2 ml AZM-free supplemented DMEM at 37 °C for 48 h. Virus titers in the culture medium and virus matrix protein 1 (M1) gene expression levels in the cells were examined by virus plaque assays and qPCR analysis, respectively.
Viral plaque assay
A confluent monolayer of MDCK cells was infected with serial dilutions of the culture medium collected from each experiment at 35 °C for 1 h. After removal of the inoculum, the cells were washed with PBS and overlaid with Eagle’s minimal essential medium (EMEM) containing 0.8% agarose, 40 mM HEPES, 0.15% sodium bicarbonate, 2 mM l-glutamine, 2 µg ml−1 trypsin, and 50 µg ml−1 gentamicin. After incubation at 37 °C for 48 h, the cells were fixed with 10% formaldehyde, followed by staining with 0.1% crystal violet solution to count viral plaques.
Half-maximal inhibitory concentration
The half-maximal inhibitory concentration (IC50) of AZM, with respect to viral proliferation, was evaluated by the procedure (iv; at the time of infection) mentioned in the “Different azithromycin treatments against virus infection in host cells” section. The viruses were premixed with 300 μl non-supplemented DMEM containing various concentrations of AZM (up to 600 μM), and the host A549 cells were immediately infected for 1 h at 35 °C. Then, the cells were washed with PBS and cultured in AZM-free supplemented DMEM for 48 h at 37 °C. Progeny virus titers in the culture medium were examined by virus plaque assays to calculate IC50 values.
Cytotoxicity assay
The cytotoxicity of AZM toward A549 cells was determined by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assays based on the manufacturer’s instructions for the Cell Proliferation Kit I (Roche, Germany). A549 cells were incubated in 300 μl non-supplemented DMEM containing various concentrations of AZM (up to 600 μM) in the presence or absence of viruses (1 MOI) for 1 h at 35 °C. After treatment, the cells were washed with PBS and cultured in AZM-free supplemented DMEM for 48 h at 37 °C. The culture medium was removed and 1 ml DMEM containing MTT labeling reagent (0.5 mg ml−1) was supplied and incubated for 3 h. Subsequently, 1 ml solubilization solution was added and incubated for 14 h at 37 °C. The solubilized formazan products were spectrophotometrically measured using an iMark microplate reader (BioRAD, USA).
Examination of inhibitory effects of AZM on budded progeny viruses
A549 cells were first infected with A(H1N1)pdm09 virus (1 MOI) at 35 °C for 1 h and cultured with or without AZM (200 µM). After 10 h of culturing, virus gene expression in the cells and titers of budded progeny viruses in the medium were measured by qPCR and plaque assays, respectively. The newly prepared A549 cells were infected with the collected culture medium containing progeny viruses with or without AZM at 35 °C for 1 h. After infection, the cells were cultured in AZM-free supplemented medium at 37 °C for 7 h and were subjected to M1 expression analysis.
Hemagglutination inhibition assay
Fresh 1% red blood cells (RBCs) in PBS solution were prepared from chicken whole blood (Biotest Company, Japan). Twenty-five microliters of serially diluted A(H1N1)pdm09 virus solution [640 hemagglutination units (HAU) ml−1] was incubated with an equal volume of PBS or AZM/EtOH in PBS solution for 30 min at room temperature (20–22 °C). Next, 50 µl of 1% RBC solution was added, followed by incubation for 20 min at room temperature. Hemagglutination was observed to estimate whether AZM inhibits the binding of virus hemagglutinin (HA) and sialic acid (SA) on RBCs.
Inhibitory assay to determine the effect of AZM on virus attachment or internalization during infection
Attachment stage assay: A549 cells were incubated with a mixture of 200 µM AZM and viruses (1 MOI) at 4 °C for 1 h. After removal of the mixture, the host cells were washed with cold PBS, and total RNA was extracted. Internalization stage assay: A549 cells were incubated with viruses (1 MOI) at 4 °C for 1 h. The cells were washed with warm PBS and cultured with medium containing 200 µM AZM at 37 °C for 1 h. After incubation, the cells were washed with PBS and treated with proteinase K (Wako, Japan) in PBS at a final concentration of 100 µg ml−1 at 37 °C for 5 min to remove viruses remaining at the cell surface. The extracted total RNA was synthesized into cDNA, and the expression levels of virus M1 and nucleoprotein (NP) were analyzed by qPCR.
Quantitative real-time PCR
One microgram of total RNA was reverse-transcribed into cDNA in a 20-µl reaction mixture using ReverTra Ace qPCR RT Master Mix with gDNA remover (Toyobo, Japan). The prepared cDNA was used for virus gene expression analyses by qPCR with PowerUp SYBR green PCR Master Mix (ThermoFisher Scientific, USA). PCR was performed using a specific primer set (Supplementary Table 1) according to the following cycles: 50 °C for 2 min, 95 °C for 2 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min.
Mice
The animal protocols of influenza virus infection were approved by the Institutional Animal Use and Care Committee and conformed to the guidelines of Teikyo University (AUP No. 16-021). Wild-type 8-week-old BALB/c female mice were purchased from SLC (Shizuoka, Japan) and housed in pathogen-free conditions.
Animal infection experiment and administration of AZM
AZM was dissolved in EtOH and mixed with PBS (pH 7.0) to prepare a solution containing 200 μg AZM in total 50-μl volume. The final concentration of EtOH in the mixture was adjusted within 3%. Anesthetized mice were intranasally infected with 300 pfu of mouse-adapted influenza A(H1N1)pdm09 virus. Six hours post-infection, whole lung tissues from one group were sampled as reference control (without treatment). Other groups were administered the mixture solution intranasally with or without AZM (10 mg kg−1) twice per day every 12 h under isoflurane anesthesia for 3 days post-infection. The rectal temperature and body weight of mice were monitored. At different time points, whole lung tissues were collected from the treated mice and homogenized in RNAiso plus solution using a beads cell disrupter (Micro SmashTM MS-100, Tomy, Japan). The cDNA pools were synthesized from the extracted total RNA, and viral M1 and NP gene expression levels were investigated by qPCR.
Statistical analyses
All experimental data were statistically analyzed by the Mann–Whitney U (MWU) test, one-way or two-way ANOVA using Graph Prism 7.02.
Results
AZM inhibits influenza A(H1N1)pdm09 virus activity by directly interacting with the viruses
To investigate the AZM treatment condition that led to the most effective antiviral activity, we performed the following experiments based on four different conditions: post-infection treatment (i), pretreatment of cells (ii), pretreatment of viruses (iii), and treatment at the time of infection (iv) using AZM. AZM administration after infection resulted in a normal progeny virus titer in the culture medium (Fig. 1a) and typical viral M1 gene expression levels in the host cells (Fig. 1b), as compared to those in the controls. Pretreating viruses with AZM for 1 h before infection resulted in a remarkable reduction in progeny virus production and M1 expression. AZM administration at the time of infection also significantly reduced progeny virus titers to similar levels observed with the pretreatment of viruses group (Fig. 1). In contrast, the pretreatment of host A549 cells with AZM for 1 h did not result in a striking difference in both progeny production and M1 expression levels compared to those in the control group (Fig. 1). The administration of clarithromycin (CAM) under these experimental conditions did not reduce progeny virus production (Suppl. Fig. 1). These observations indicated that AZM interacts with A(H1N1)pdm09 viruses to inhibit virus activity in the early phases of infection. Both 1 h pretreatment and treatment at the time of infection with AZM showed similar inhibitory effects on progeny production.

Antiviral activity of azithromycin (AZM) on A(H1N1)pdm09 virus infection. Antiviral activity of AZM was evaluated under four different conditions (i) Post-infection treatment: A549 cells were infected with the viruses before culturing with or without AZM. (ii) Pretreatment of cells: A549 cells were pretreated with or without AZM before infection. Then, the cells were cultured in AZM-free medium. (iii) Pretreatment of viruses: the viruses were pretreated with or without AZM for 1 h before infecting A549 cells, followed by culturing in AZM-free medium. (iv) At the time of infection: A549 cells were infected with the premix of viruses and AZM for 1 h, and cultured in AZM-free medium. Virus titers in culture medium (a) and viral M1 gene expression level in A549 cells (b) were examined after 48-h culture. The values of AZM-treated cells were converted as percent index and are shown by means± S.E. from six individual data. *p < 0.01, n.s., no significant differences (MWU test)
AZM exerts no cytotoxicity towards host cells in the IC50 range
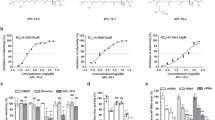
We next determined the IC50 value of AZM on progeny virus proliferation (Fig. 2). AZM decreased progeny viruses released into the culture medium in a dose-dependent manner, and the mean IC50 value was approximately 68 µM (Fig. 2a). The expression status of viral M1 gene in A549 cells correlated with the trend in virus titers (Suppl. Fig. 2).

Half-maximal inhibitory concentration (IC50) and cytotoxicity of AZM. a The IC50 of AZM on progeny virus titers in culture medium, with representative images of formed plaques (lower pictures). The graph is shown as means ± S.D. from six individual data. b Cytotoxicity of AZM on host A549 cells under non-infectious (upper panel) or infectious condition (lower panel). A549 cells were incubated with various concentrations of AZM in the absence or presence of the viruses. The cells were cultured in AZM-free medium for 48 h and subjected to MTT assay. Each graph is expressed by means ± S.D. from six individual data, *p < 0.01, one-way ANOVA
To determine the concentration at which AZM exhibits toxicity towards host A549 cells, MTT assays were performed with a broad range of AZM concentrations (Fig. 2b). Under non-infectious conditions, significant cytotoxicity was not detected by co-culturing with less than 200 μM AZM (Fig. 2b, upper panel). Similarly, no cytotoxic effect on A549 cells was observed in the presence of AZM at less than 600 μM under infectious conditions (Fig. 2b, lower panel). These data indicate that AZM does not influence host cell viability within the IC50 range in both non-infectious and infectious conditions.
AZM does not influence attachment status but affects viral internalization
To explore the mechanisms underlying the antiviral activity of AZM, we first determined whether AZM interferes with the binding interaction between HA of virus and SA on RBCs. As seen in Fig. 3, hemagglutination was observed up to a virus dilution of 1/16; however, no marked interruption of hemagglutination by AZM was detected within this dilution range, indicating that AZM did not affect the binding activity between virus HA and its SA receptor on the cells. We further investigated the inhibitory mechanism associated with the effects of AZM on virus attachment and internalization based on the expression profiles of virus genes in the host cells (Fig. 4). Treating viruses with AZM at the time of infection did not lead to changes in M1 and NP expression, which was determined from the attached viruses on the cell surface (Fig. 4a). In contrast, AZM administration after virus attachment, followed by the removal of orphan viruses using protein-K, significantly reduced both M1 and NP expression in host cells (Fig. 4b). These observations indicate that AZM does not influence binding ability, but interferes with the internalization process during the early phase of virus invasion.

Hemagglutination inhibition profile of AZM. Serially diluted virus solution was incubated with an equal volume of PBS (control) or AZM (at the indicated concentrations), respectively. Fresh 1% RBCs was added to each well, and then hemagglutination between RBCs and viruses was detected (left panel). The graph (right panel) is expressed as hemagglutination units (HAU) versus AZM concentration. Representative data from two independent experiments are shown

Effect of AZM treatment on virus attachment and internalization. Inhibitory activity of AZM at the attachment (a) or internalization stages (b) of viruses based on expression level of viral M1 (left) and NP genes (right). Attachment stage: the viruses were premixed with or without AZM. A549 cells were infected with the viruses for 1 h at 4 °C, and virus gene expression in the cells was analyzed. Internalization stage: A549 cells were infected with viruses at 4 °C for 1 h and then cultured with or without AZM at 37 °C for 1 h. After that, the cells were treated with proteinase K and subjected to gene expression analysis. Data are expressed as means ± S.E. from nine individual data by three independent experiments. *p < 0.05, n.s., no significant differences (MWU test)
AZM targets newly synthesized progeny viruses
Based on the inhibitory effect of AZM on the internalization of parental viruses during infection, we hypothesized that AZM could interrupt a repeat cycle of infection and progeny virus propagation. To prove this hypothesis, we monitored virus quantities at each point during the initial infection of parental viruses and the second infection of progeny viruses in the presence of AZM (Fig. 5). First, host A549 cells were infected with A(H1N1)pdm09 viruses; then, the cells were co-cultured with or without AZM for 10 h. At this time point, progeny virus titers in the culture medium or viral M1 expression levels in the host cells were comparable in the presence and absence of AZM (Fig. 5a, b). This result was consistent with the observations shown in Fig. 1. Subsequently, we infected newly prepared A549 cells with the collected culture supernatant, which contained budded progeny viruses, in the presence or absence of AZM. The infected A549 cells were then cultured in AZM-free medium for 7 h. At this point, M1 expression levels were remarkably reduced in A549 cells upon exposure to medium containing progeny viruses and AZM (Fig. 5c). These observations confirm our theory that AZM can prevent virus internalization when extracellular viruses invade host cells.

Inhibitory effect of AZM on progeny virus proliferation. A549 cells were first infected with viruses and co-cultured with or without AZM. After 10-h culture from the first infection, progeny virus titers in culture medium (a) and viral M1 expression in host cells (b) was examined. Harvested medium containing budded progeny viruses as well as AZM was exposed to newly prepared A549 cells. The cells were cultured in AZM-free condition for 7 h, and then M1 gene expression in the cells was analyzed (c). Data are expressed as means ± S.E. of 12 individual data from three independent experiments. *p < 0.05, n.s., no significant differences (MWU test)
Single administration of AZM relieves viral load in infected mice
Considering our in vitro observations, we next perform intranasal administration of AZM for in vivo challenge (Fig. 6a). As shown in Fig. 6b, AZM administration tended to reduce viral M1 and NP expression in the lung tissues 3 days after infection. The maximal inhibition of viral expression was observed 2 days post-infection, when the viruses propagated dramatically (Fig. 6b). Further, AZM treatment alleviated the decrease in body temperature 3 days after infection (Fig. 6c), but had no effect on body weight in infected mice at any day (Fig. 6d). Our in vivo challenge showed that a single treatment with AZM via the intranasal route could suppress the virus load in the lungs, thereby preventing hypothermia during A(H1N1)pdm09 virus infection.

Therapeutic advantages of AZM on mice. a Schematic procedure is as follows: all mice were intranasally infected with mouse-adapted viruses. After inoculation, lung tissues dissected from the non-administered group was collected as a reference control. Other groups were intranasally administered with or without AZM twice a day for 3 days. At the indicated time points, lung tissues were sampled from the treated mice. b Expression of viral M1 (left) and NP gene (right) in the lungs. Each gene expression level was normalized by that of GAPDH and relatively compared between control and AZM-administered groups at 1, 2, and 3 days post-infection based on reference control. The graphs are shown by median, with an interquartile range from more than five individual data (control: n = 5 and AZM: n = 5–6 each day). *p < 0.05, **p < 0.01 (two-way ANOVA). Actual body temperature (c) and percent body weight (d) of infected mice. Each vital sign was monitored and compared between control and AZM-treated mice during before and after the infection. Data are shown as median with an interquartile range from indicated individual mouse. ***p < 0.001 (two-way ANOVA)
Discussion
In this study, we aimed to determine the anti-influenza A(H1N1)pdm09 virus activity of AZM and to elucidate the underlying mechanism. We found that AZM exerts anti-influenza A(H1N1)pdm09 virus activity based on both in vivo and in vitro studies. The administration of AZM after infection did not inhibit progeny virus replication, whereas AZM treatment before infection remarkably reduced progeny virus production after 48 h of culture. We also showed that existing AZM in the culture medium interfered with the infection activity of budded progeny viruses. These in vitro observations indicate that AZM inhibits influenza A virus activity, and its antiviral activity is effective when the viruses are located outside host cells during a repeat cycle of propagation. AZM administration had no effect on progeny titer after infection, implying that it cannot block progeny virus yield. AZM is therefore capable of interfering with virus entry into host cells during the early phase of the infection process. The infection of influenza A viruses is generally established through the binding of viral HA and SA on the cell surface [18]. In our study, AZM did not affect this binding on the host cell surface (Figs. 3 and 4a). In contrast, AZM significantly affected virus internalization, which is the second stage of virus invasion (Fig. 4b). The internalization of influenza A viruses is accomplished by endocytosis. Virus ribonucleoproteins (vRNPs) are de-enveloped, which depends on the acidified environment of endosomes and released into the cytoplasm, which is followed by the initiation of component multiplication for progeny virus replication [19]. Several macrolides such as CAM and bafilomycin A1 (Baf-A1) attenuate the propagation of influenza A/PR/8/34(H1N1) and A(H3N2) viruses, respectively, by impairing the formation of acidic endosomes in host cells [16, 20]. The endocytosis of influenza A viruses is mainly mediated by clathrin-associated molecules [21], which are host cell factors. Pretreating host A549 cells with AZM before infection did not inhibit progeny virus production in our study. This indicates that AZM does not affect host factors including clathrin-associated molecules to induce antiviral effects.
In contrast, treating viruses with AZM before infection decreased the quantity of internalized viruses in host cells (Fig. 5c). It takes more than 30 min for vRNPs to be uncoated and released into the cytoplasm [22]. In our internalization assay, the treated host cells were promptly harvested to avoid amplification of virus nucleotide copies in the cells. Further, blockage of vRNP uncoating by AZM is unlikely, because the total quantity of virus RNA is encased inside cells regardless of whether endocytosed viruses undergo uncoating. Based on these in vitro observations, we suggest that AZM-pretreated viruses attach normally to the cell surface; however, more than half of the viruses could not internalize into the cells and remained at the cell surface. Our data indicate the possibility that AZM acts directly on the influenza virus, and that the treated viruses cannot internalize into host cells. Moreover, AZM had no effect on binding between SA and HA; nonetheless, it interfered with virus internalization. This suggests that alternative receptor(s) containing are involved in virus endocytosis. The entry of influenza A viruses into cells is mediated by interactions with lectin receptors, independent of the SA–HA interaction pathway [23]. It is possible that AZM hampers the interaction between the virus and such receptor(s) to prevent internalization.
Pretreating neither A(H1N1)pdm09 viruses nor host cells with CAM inhibited progeny virus production (Suppl. Fig. 1), whereas AZM interrupted internalization in this study. CAM inhibits A(H1N1) PR8 virus activity [15, 16], but it did not affect A(H1N1)pdm09 virus proliferation. These observations indicate the possibility that the unique anti-influenza virus mechanism of AZM is fundamentally different from that of CAM.
One in vivo study reported that the intraperitoneal injection of AZM (100 mg kg−1, one dose) at 48 h post-infection could reduce virus titers in the lung until death [24]. In that study, the additional oral administration of oseltamivir was more efficient in suppressing the virus titer, leading to a survival advantage. In contrast, we selected the intranasal administration of AZM from the initial phase of infection (10 mg kg day−1), and this route significantly reduced viral loads in the lungs, in addition to providing relief from infection-induced hypothermia. Thus, the inhalation treatment of AZM concomitant with the oral administration of oseltamivir might offer better clinical benefits as a combination therapy for influenza virus infection.
A(H1N1)pdm09 virus is currently a seasonal influenza that causes annual epidemic outbreaks. As a licensed anti-influenza drug, laninamivir is clinically administered via the inhalation route to humans. AZM is also a safe and licensed drug, and accordingly, it showed low cytotoxicity under both non-infectious and infectious conditions in this study. The therapeutic benefits of intranasal AZM in mice infected with A(H1N1)pdm09 virus provide a new therapeutic perspective to deal with seasonal influenza epidemics. Influenza A viruses that are resistant to neuraminidase and M2 channel inhibitors such as oseltamivir and amantadine have emerged recently in Japan [25, 26]. Therefore, the continual development and/or repositioning of anti-influenza virus agents is of importance to public health. In this study, we show the potential of AZM to exert antiviral activities both before and after influenza A virus infection, suggesting that it has potential for prophylactic administration. As AZM is an antibiotic that possesses anti-bacterial activity, its continuous use poses a risk for the emergence of anti-bacterial resistance. However, no casualties were observed in patients who progressed to respiratory tract complications caused by secondary bacterial infection in clinical practice [27]. Thus, AZM could be prescribed to prevent both primary infection by influenza A virus and secondary infection by bacteria. Therefore, the anti-bacterial activity of AZM is not necessarily associated with shortcomings for its clinical use against human influenza. Owing to their unique chemical architecture, macrolides exert anti-bacterial and antiviral activities independently. The erythromycin-based derivative EM900 inhibits several viruses including influenza A virus [17, 28]. Some AZM-derivatives synthesized by our group showed anti-AH1N1pdm09 virus activity with less potent anti-bacterial activity (data not shown). Thus, the different components of the chemical architecture responsible for anti-influenza A virus activity should be investigated to facilitate the development of optimal anti-influenza drugs based on macrolides. Further in vitro investigations, for example, to determine whether AZM is directly involved in particular region(s) on A(H1N1)pdm09 virus for inactivation, are necessary to understand the detailed anti-influenza virus mechanism of AZM. In addition, to ascertain the consequences of intranasal AZM treatment in vivo, we must perform follow-up experiments such as assessing survival in lethally infected mice. However, the findings of this study could form the basis of the repositioning of this anti-influenza drug for widespread clinical treatment options for human influenza.
References
Glezen WP, Taber LH, Frank AL, Gruber WC, Piedra PA. Influenza virus infections in infants. Pediatr Infect Dis J. 1997;16:1065–8.
Tumpey TM. et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310:77–80.
Tran TH. et al. Avian influenza A (H5N1) in 10 patients in Vietnam. N Engl J Med. 2004;350:1179–88.
World Health Organization. Weekly update pandemic (H1N1) 2009. 6 August 2010. https://www.who.int/csr/don/2010_08_06/en/.
Mauad T. et al. Lung pathology in fatal novel human influenza A (H1N1) infection. Am J Respir Cit Care Med. 2009;181:72–9.
Takeda S. Extracorporeal membrane oxygenation for 2009 influenza A (H1N1) severe respiratory failure in Japan. J Anesth. 2012;26:650–7.
Hayden FG. et al. Efficacy and safety of the neuraminidase inhibitor zanamivir in the treatment of influenza virus infections. GG167 Influenza Study Group. N Engl J Med. 1997;337:874–80.
Yamashita M. Laninamivir and its prodrug, CS-8958: long-acting neuraminidase inhibitors for the treatment of influenza. Antivir Chem Chemother. 2010;21:71–84.
Stephenson I. et al. Neuraminidase inhibitor resistance after oseltamivir treatment of acute influenza A and B in children. Clin Infect Dis. 2009;48:389–96.
Hurt AC, Holien JK, Parker M, Kelso A, Barr IG. Zanamivir-resistant influenza viruses with a novel neuraminidase mutation. J Virol. 2009;83:10366–73.
Leang SK. et al. Peramivir and laninamivir susceptibility of circulating influenza A and B viruses. Influenza Other Respir Virus. 2014;8:135–9.
Furuta Y, Komeno T, Nakamura T. Favipiravir (T-705), a broad-spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B. 2017;93:449–63.
Kimberly ENg. Xofluza (Baloxavir Marboxil) for the treatment of acute uncomplicated influenza. P T Community. 2019;44:9–11.
Takashita E. et al. Detection of influenza A (H3N2) viruses exhibiting reduced susceptibility to the novel cap-dependent endonuclease inhibitor baloxavir in Japan, December 2018. Eur Surveill. 2019;24:1800698
Tsurita M. et al. Early augmentation of interleukin (IL)-12 level in the airway of mice administrated orally with clarithromycin or intranasally with IL-12 results in alleviation of influenza infection. J Pharm Exp Ther. 2001;298:362–8.
Yamaya M. et al. Clarithromycin inhibits type A seasonal influenza virus infection in human airway epithelial cells. J Pharm Exp Ther. 2010;333:81–90.
Sugamata R. et al. Leucomycin A3, a 16-membered macrolide antibiotic, inhibits influenza A virus infection and disease progression. J Antibiot. 2014;67:213–22.
Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–69.
Palese P, Shaw ML. Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields virology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2007. p. 1647–89.
Yeganeh B. et al. Suppression of influenza A virus replication in human lung epithelial cells by noncytotoxic concentrations bafilomycin A1. Am J Physiol Lung Cell Mol Physiol. 2015;300:L270–86.
Le Roy C, Wrana JL. Clathrin- and non- clathrin-mediated endocytosis regulation of cell signaling. Nat Rev Mol Cell Biol. 2005;6:112–26.
Qin J. et al. Real-time dissection of dynamic uncoating of individual influenza viruses. Proc Natl Acad Sci USA. 2019;116:2577–82.
Londrigan SL. et al. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. J Virol. 2011;85:2990–3000.
Fage C, Pizzorno A, Rheaume C, Abed Y, Boivin G. The combination of oseltamivir with azithromycin does not show additional benefits over oseltamivir monotherapy in mice infected with influenza A(H1N1)pdm2009 virus. J Med Virol. 2017;89:2239–43.
Matsuzaki Y. et al. A two-year survey of the oseltamivir-resistant influenza A(H1N1) virus in Yamagata, Japan and the clinical effectiveness of oseltamivir and zanamivir. Virol J. 2010;7:53
Dong G. et al. Adamatane-resistant influenza A viruses in the world (1902-2013): frequency and distribution of M2 gene mutations. PloS ONE. 2015;10:e0119115
Louie J. et al. Factors associated with death or hospitalization due to pandemic 2009 influenza A(H1N1) infection in California. JAMA. 2009;302:1896–902.
Kalonji L. et al. The non-antibiotic macrolide EM900 inhibits rhinovirus infection and cytokine production in human airway epithelial cells. Physiol Rep. 2015;3:e12557
Acknowledgements
This study was supported by Japan Agency for Medical Research and Development (AMED) under Grant number: 16jm0210032h0004 and Japan Science and Technology Agency (JST). We appreciate Dr. Yosuke Kameoka at the A-CLIP institute for providing the A(H1N1)pdm09 virus strain. We would also like to thank Editage (www.editage.jp) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Tran, D.H., Sugamata, R., Hirose, T. et al. Azithromycin, a 15-membered macrolide antibiotic, inhibits influenza A(H1N1)pdm09 virus infection by interfering with virus internalization process. J Antibiot 72, 759–768 (2019). https://doi.org/10.1038/s41429-019-0204-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-019-0204-x
This article is cited by
-
Rapid and sensitive detection of SARS-CoV-2 virus in human saliva samples using glycan based nanozyme: a clinical study
Microchimica Acta (2024)
-
Unnatural activities and mechanistic insights of cytochrome P450 PikC gained from site-specific mutagenesis by non-canonical amino acids
Nature Communications (2023)
-
Assessment of drugs administered in the Middle East as part of the COVID-19 management protocols
Inflammopharmacology (2022)
-
A motley of possible therapies of the COVID-19: reminiscing the origin of the pandemic
Environmental Science and Pollution Research (2022)
-
Current trends in diagnosis and treatment strategies of COVID-19 infection
Environmental Science and Pollution Research (2021)
